La lettre Surrénale – Octobre 2012
 |
||||||||
|
||||||||
|
||||||||
|
Une nouvelle lettre pour apprivoiser les surrénales Bienvenue à la première “Newsletter” consacrée aux glandes surrénales ! Ces petites glandes causent souvent des soucis à l’endocrinologue : pathologies rares, dont les explorations peuvent être complexes… et les complications gravissimes : pas question de les ignorer. Avec cette newsletter, nous souhaitons mieux faire connaître les différents aspects de leurs pathologies, autant sur les plans physiopathologique que clinique, et nous souhaitons tenir le lecteur informé des avancées aussi bien dans le domaine de la corticosurrénale que dans celui de la médullosurrénale et du paragangliome. Dans ce premier numéro, nous avons fait appel à des experts cliniciens et scientifiques (Éric Baudin, Hervé Lefebvre, Michael Thomas) ainsi qu’à une plus jeune (Marie-Aude Quemerais) pour présenter les nouveautés dans le traitement du corticosurrénalome (E.B., M.T.), le risque d’insuffisance surrénale aiguë dans l’hyperplasie congénitale (M.A.-Q.) et la physiopathologie moléculaire de l’insuffisance surrénale (H.L.). Enfin, votre serviteur a essayé de faire le point sur les derniers gènes des phéochromocytomes et paragangliomes. Bonne lecture ! Nous espérons bien que vous en redemanderez ! |
||||||||
 |
||||||||
|
Corticosurrénalome malin : FIRM-ACT établit le premier standard de polychimiothérapie L’étude FIRM-ACT, premier essai international de phase 3 dans le corticosurrénalome, a permis de définir le premier standard de polychimiothérapie de cette pathologie dont le pronostic reste sombre. Cet essai montre qu’il est possible de mettre en place des études randomisées chez des patients porteurs d’une tumeur rare, grâce à l’implication de centres experts européens réunis en réseaux. Dans sa pratique quotidienne, l’endocrinologue est confronté aux cancers endocrines, parmi lesquels 3 formes sont particulièrement graves : le carcinome anaplasique de la thyroïde, le carcinome neuroendocrine peu différencié et le corticosurrénalome malin. Ces 3 cancers, outre leur gravité, partagent leur rareté et on a longtemps pensé qu’il était impossible de mettre en place des études randomisées pour définir des standards thérapeutiques. L’incidence du corticosurrénalome, qui se développe aux dépens du cortex surrénalien, est estimée entre 1 et 2 cas par million d’habitants. La survie à 5 ans est en moyenne de 50 %, mais elle est inférieure à 15 % au stade métastatique. La prise en charge thérapeutique repose en priorité sur la chirurgie d’exérèse. FIRM-ACT a montré un bénéfice cliniquement significatif de l’association MEDP versus MS, avec une survie globale (SG) de respectivement 14,8 mois versus 12 mois, une survie sans progression de 5 mois versus 2 mois et un taux de réponse objective de 23 % versus 9 %. Les taux de complications ont été comparables dans les 2 groupes. L’étude n’a pas permis de mettre en évidence de facteurs prédictifs de réponse. Au total l’étude FIRM-ACT permet d’établir un premier standard de chimiothérapie dans le corticosurrénalome malin : l’association MEDP. Elle montre aussi qu’il est possible de réaliser une étude randomisée dans une pathologie aussi rare que le corticosurrénalome. Cet effort majeur a été rendu possible grâce à la motivation de centres experts européens, regroupés au sein de réseaux nationaux et internationaux, dont le réseau INCa-COMETE (Institut National du Cancer-COrtico-MEdullo-Tumeurs Endocrines) en France. Des progrès restent bien sûr à accomplir, comme l’atteste la médiane de SG des patients, de 15 mois dans le meilleur bras. L’optimisation de ce nouveau standard constitue une voie de recherches futures, et cette première étude doit également permettre d’envisager d’autres collaborations de ce type pour évaluer des molécules innovantes. En termes de nouvelles approches thérapeutiques, les résultats de l’étude OSI 906, deuxième étude de phase 3 internationale dans le corticosurrénalome malin, évaluant un inhibiteur du récepteur de l’IGF1 versus placebo, sont attendus. Référence bibliographique …………………………………………………………………………………………………………………………………………………………… Récepteur alpha 2 de l’interleukine 13 : une nouvelle cible pour le traitement du corticosurrénalome La prévalence des carcinomes de la corticosurrénale dans la population générale est très faible, avec seulement 1 à 12 cas par million chez l’adulte (1). Les mécanismes moléculaires impliqués dans leur développement sont mal connus. Les études du patron d’expression des gènes ont largement été utilisées dans un but de classification et de pronostic de la malignité de ces tumeurs (2, 3). L’intérêt de l’article publié par E. Kebebew et al. (4) réside dans l’utilisation des données du transcriptome dans un but thérapeutique. En effet, dans une étude précédente (5), le même groupe avait identifié 37 gènes dont le profil d’expression différait selon que les tumeurs étaient bénignes ou malignes. Parmi ceux-ci, le gène codant pour le récepteur alpha2 de l’interleukine-13 (IL-13R?2) était retrouvé surexprimé dans les carcinomes. Ainsi le ciblage de récepteurs de surface pouvait être une option intéressante pour le développement de thérapies ciblées et c’est cette voie que les auteurs ont poursuivie. Le ligand naturel de ce récepteur est l’interleukine-13 (IL-13), les auteurs montrent qu’une protéine de fusion qui combine IL-13 et la forme mutée de l’exotoxine de Pseudomonas (IL-13-PE) est cytotoxique pour les cellules tumorales de diverses pathologies exprimant IL-13R?2. Par des approches in vitro et in vivo, ils montrent également que l’inhibition de l’expression ou le blocage de l’IL-13R?2 dans les cellules NCI-H295R entraînent une diminution de leur capacité d’invasion, de prolifération, de synthèse de protéines et de croissance sous la peau de souris immunodéficientes. |
||||||||
 |
||||||||
|
Insuffisance surrénale aiguë dans les hyperplasies congénitales des surrénales par déficit en 21-OH hydroxylase Chez les patients porteurs d’une hyperplasie congénitale des surrénales, la fréquence de l’insuffisance surrénale aiguë est de l’ordre de 6 cas pour 100 patients/année. Quels sont les facteurs de prédisposition et comment la prévenir ? L’hyperplasie congénitale des surrénales par déficit en 21-hydroxylase (HCS) est une maladie autosomique récessive dont l’incidence est estimée à 1/11 000 naissances). Ce déficit aboutit à un défaut de production du cortisol, à une sécrétion excessive d’androgènes surrénaliens et, dans 2/3 des cas, à un déficit de sécrétion d’aldostérone (formes avec “perte de sel”). Le risque d’insuffisance surrénale aiguë (ISA) doit être prévenu, la vie durant, par un ajustement adéquat des doses de glucocorticoïdes en situation de stress. L’objectif de cette étude rétrospective (1) est donc de déterminer la fréquence et les causes de l’ISA dans la population des patients ayant une HCS, en se fondant sur 2 analyses complémentaires : 122 questionnaires spécifiques remplis par des patients suivis à Munich, Würzburg et Berlin, ou par des membres de l’association allemande d’entraide des HCS (âge moyen : 35 ans), et 67 dossiers médicaux de patients suivis à l’hôpital universitaire de Munich (âge moyen : 31 ans). La fréquence de l’ISA est, d’après les questionnaires, de 5,8 cas pour 100 patients/année et, d’après les dossiers médicaux, de 4,9 cas pour 100 patients/année. En excluant les primodécompensations (la majorité des ISA avant 2005, année de l’instauration d’un dépistage néonatal systématique), cette fréquence reste, respectivement, de 4,9 et de 3,8 cas pour 100 patients/année. Une fréquence d’ISA discrètement supérieure (6,6 cas pour 100 patients/année) était observée dans le groupe de patients insuffisants surrénaux primaires d’une autre étude allemande récente (2). Cette différence s’explique, selon N. Reisch et al. (1) , par une survenue beaucoup plus précoce, dès l’enfance, de l’HCS et, de ce fait, par une éducation plus répétée et efficace quant aux mesures de prévention. Une autre explication (3) pourrait être une amélioration partielle du déficit minéralocorticoïde par la 21-hydroxylation extrasurrénalienne des précurseurs en excès (ex. : CYP2C19 et CYP3A4 hépatiques capables d’hydroxyler la progestérone). Les facteurs déclenchants retrouvés dans les dossiers médicaux sont dans 29 % des cas une gastroentérite aiguë et dans 18 % des cas une crise avec perte de sel, 14 % des ISA restant inexpliquées. La fréquence de survenue ne dépend pas du sexe mais de l’âge, avec plus de 70 % d’ISA les 10 premières années de vie (1/3 la première année). À noter un deuxième pic de survenue après la puberté (18-25 ans), où le patient se gère seul sans l’aide de ses parents ; il s’agit d’une période à risque de mauvaise observance : une très bonne collaboration entre pédiatres et endocrinologues est alors nécessaire pour assurer la transition. Au final, les auteurs soulignent 4 facteurs impliqués dans la survenue d’ISA et autant de stratégies de préventions : Références bibliographiques |
||||||||
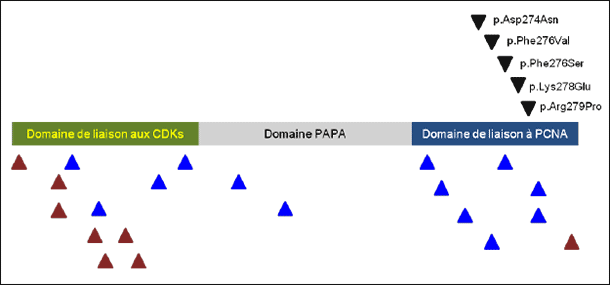
|
…………………………………………………………………………………………………………………………………………………………… Quand une mauvaise empreinte laisse une mauvaise IMAGe Une kinase dépendante des cyclines est impliquée à la fois dans le corticosurrénalome du syndrome Beckwith-Wiedemann et dans l’insuffisance surrénale du syndrome IMAGe. La prolifération cellulaire est contrôlée par des mécanismes subtils faisant intervenir de multiples complexes protéiques nucléaires. En particulier, l’entrée dans le cycle cellulaire est favorisée par les kinases dépendantes des cyclines (Cyclin-Dependent Kinases [CDK]) et est freinée par une autre famille de protéines, les inhibiteurs de CDK (CDK inhibitors [CDKI]), capables de se lier au site catalytique des CDK et d’inactiver ainsi leur activité. Parmi ces dernières, p57KIP2, encore appelée Cyclin-Dependent Kinase Inhibitor 1C (CDKN1C), est un puissant répresseur du cycle cellulaire, l’hyperexpression forcée de p57KIP2 conduisant à l’arrêt de la prolifération cellulaire en phase G1 (1, 2). Comme d’autres CDKI, la protéine CDKN1C contient plusieurs domaines fonctionnels avec notamment, dans sa région N-terminale, le domaine de liaison aux CDK et, au niveau C-terminal, un domaine de liaison au PCNA (Proliferating Cell Nuclear Antigen), une protéine impliquée dans la réplication de l’ADN (figure1) [3, 4]. Le gène CDKN1C, situé en 11p15, est soumis à empreinte parentale ; dans la plupart des tissus, l’allèle paternel est inactivé, l’expression de la protéine résultant essentiellement de la traduction de l’allèle maternel. De façon intéressante, il avait été noté que CDKN1C était fortement exprimé dans la glande surrénale (5) et, à la fin des années 1990, un groupe japonais avait pu montrer que le syndrome de Beckwith-Wiedemann, qui s’associe chez l’enfant à un risque élevé de corticosurrénalome malin, pouvait être causé par des mutations touchant l’allèle maternel du gène CDKN1C (6, 7). Il s’agit à la fois de mutations non-sens aboutissant à la perte quasi complète de la protéine et de mutations faux-sens affectant principalement le domaine de liaison aux CDK (figure1) [6, 8]. Par ailleurs, en accord avec les données issues de l’observation des cas de syndrome de Beckwith-Wiedemann, le KO (knock-out) de CDKN1C chez la souris entraîne l’apparition d’une hyperplasie surrénalienne (9). L’ensemble de ces résultats suggérait donc que CDKN1C pouvait jouer un rôle important dans la tumorigenèse surrénalienne, la répression de son expression favorisant une hyperactivité des complexes cyclines-CDK et, consécutivement, l’emballement du cycle cellulaire. Des données complémentaires, rapportées notamment par l’équipe de Christine Gicquel à l’hôpital Trousseau, sont venues conforter cette hypothèse en montrant que l’expression de CDKN1C était fortement inhibée dans les corticosurrénalomes malins, par opposition aux tumeurs corticosurrénaliennes bénignes dont le niveau d’expression est comparable à celui de la surrénale normale (10). Dans les tumeurs malignes, le déficit d’expression de CDKN1C, qui semble donc se comporter comme un gène suppresseur de tumeurs, paraît provenir d’une perte de l’allèle maternel avec duplication de l’allèle paternel ou d’anomalies de l’empreinte génétique (10). Le syndrome IMAGe est une affection exceptionnelle rapportée pour la première fois en 1999 (11). Il associe un retard de croissance intra-utérin, une dysplasie métaphysaire, diverses anomalies génitales et une hypoplasie surrénalienne responsable d’une insuffisance surrénale sévère en période postnatale précoce (11). Sa présentation peut être sporadique ou familiale (12). É. Vilain et al., à Los Angeles (États-Unis), viennent de montrer, grâce à l’étude d’un cas familial et de cas sporadiques, que le syndrome IMAGe est lui aussi la conséquence de mutations faux-sens de CDKN1C (13). Les 5 mutations hétérozygotes décrites siègent toutes dans le domaine de liaison au PCNA (figure1). L’expression phénotypique des mutations n’est présente que lorsque l’allèle maternel est touché. Grâce à des approches expérimentales très élégantes, utilisant à la fois des modèles in vivo et in vitro, les auteurs ont pu montrer que, contrairement aux mutations rencontrées au cours du syndrome de Beckwith-Wiedemann, qui réduisent la capacité de CDKN1C à bloquer le cycle cellulaire en phase G0/G1, les altérations génétiques observées chez les patients atteints du syndrome IMAGe n’affectaient pas l’effet inhibiteur de la protéine sur la progression du cycle cellulaire (13). À l’inverse, ces mutations aboutissent à un gain de fonction de CDKN1C responsable d’une freination de la prolifération cellulaire et d’une hypoplasie tissulaire. La forte expression de CDKN1C dans la glande surrénale fœtale, en particulier dans la zone sous-capsulaire qui contient les cellules souches surrénaliennes, pourrait expliquer pourquoi la surrénale est particulièrement touchée par l’hypoplasie tissulaire au cours du syndrome IMAGe (13). É. Vilain et al. ont également montré que les mutations de CDKN1C rencontrées au cours du syndrome IMAGe retentissaient négativement sur sa capacité à lier PCNA, ce qui aboutit à un défaut d’ubiquitination de la protéine, anomalie probablement impliquée dans son gain de fonction (13). La démonstration de l’implication majeure de p57KIP2/CDKN1C dans le développement et la physiopathologie surrénalienne vient donc d’être grandement renforcée par l’identification de son rôle dans la pathogénie du syndrome IMAGe. Cette découverte va certainement susciter de multiples travaux portant notamment sur le rôle de la protéine au cours du développement surrrénalien et sur ses interactions éventuelles avec les facteurs transcriptionnels connus pour être impliqués dans la formation et la différenciation du cortex surrénal, tels que SF1, DAX1 et les facteurs GATA. On peut également imaginer que la protéine p57KIP2/CDKN1C est impliquée dans les hypoplasies surrénaliennes acquises, comme celles que l’on rencontre au cours de la maladie d’Addison auto-immune. Enfin, une voie originale du traitement des corticosurrénalomes malins pourrait être de tenter de faire réexprimer la protéine dans les tumeurs et ainsi de freiner leur expansion, ce qui impliquerait au préalable de bien comprendre les mécanismes gouvernant son expression dans les cellules corticosurrénaliennes. Pour conclure, il est clair que la saga de p57KIP2/CDKN1C dans la glande surrénale est loin d’être terminée. De nouveaux développements sont à attendre, qui pourraient bien modifier profondément notre compréhension des maladies surrénaliennes et, à terme, notre arsenal thérapeutique pour la prise en charge des patients atteints de pathologies de la glande surrénale. Figure 1:
Références bibliographiques |
||||||||
|
…………………………………………………………………………………………………………………………………………………………… Les 10 gènes des phéochromocytomes et paragangliomes Deux nouveaux gènes sont impliqués dans les phéochromocytomes et paragangliome : TMEM-27 et MAX, par ailleurs NF1 joue un rôle beaucoup plus important que prévu. Le phéochromocytome était traditionnellement connu comme la “tumeur des 10 %” : 10 % de cas malins, 10 % de cas extrasurrénaliens (appelés alors paragangliomes) enfin 10 % de cas d’origine familiale, les gènes connus alors étant RET (néoplasie endocrinienne multiple de type 2), VHL (maladie de von Hippel-Lindau) et NF1 (neurofibromatose de type 1 ou maladie de von Recklinghausen). En fait, la barrière des 10 % fait partie de l’histoire : il y a déjà 10 ans, elle était allègrement franchie avec la découverte de mutations des gènes SDH (succinate déshydrogénase) chez des patients porteurs de phéochromocytomes et paragangliomes familiaux : SDHD (paragangliomes surtout cervicaux, rarement malins, transmission presque exclusivement maternelle), SDHB (paragangliomes surtout abdominaux et thoraciques, haut risque de malignité) puis, plus rarement, SDHC, SDHA et SDHAF2, soit déjà 8 gènes responsables au total non plus d’un dixième, mais plutôt d’un tiers des cas de phéochromocytomes et de paragangliomes. Ces 2 dernières années, 2 nouveaux gènes : TMEM127 (1) et MAX (2) permettent de retrouver le chiffre 10. Le gène TMEM127 code pour une protéine à 3 domaines transmembranaires dont la fonction, encore mal connue, comprend une inhibition de la voie de mTOR (1). Très récemment, l’équipe de l’hôpital européen Georges-Pompidou (HEGP), dirigée par Anne-Paule Gimenez-Roqueplo, a pu préciser la fréquence de mutations germinales de TMEM127 dans une cohorte de 642 patients porteurs de phéochromocytomes ou de paragangliomes (3). Ces mutations restent rares : elles concernaient 6 patients, soit 0,90 %, dont 3 avaient une présentation suggestive de forme génétique (jeune âge, phéochromocytome bilatéral, histoire familiale) alors que les 3 autres présentaient un phéochromocytome sporadique “banal”.
Quels messages dégager de cette avalanche de découvertes génétiques ? Tout d’abord sur le plan physiopathologique, une étude systématique par microarray des voies activés par ces gènes a permis de dégager 2 mécanismes cellulaires à l’œuvre dans les phéochromocytomes et/ou les paragangliomes : la voie de signalisation de l’hypoxie (gènes VHL et SDH) et une voie qui implique différents récepteurs protéines kinases (RET, NF1, TMEM127 et MAX). Sur le plan clinique, cette division se traduit par une sécrétion préférentielle soit de noradrénaline pour le premier groupe, soit d’adrénaline pour le second (avec un phénotype intermédiaire pour les mutations MAX). Bien sûr, ces découvertes ont d’abord un intérêt essentiel pour les patients porteurs de phéochromocytomes et leur famille : l’identification d’une mutation de 1 des 10 gènes va permettre de mieux préciser les risques de récidive, de malignité, et d’apparition de lésions associées chez un cas index, et elle sera essentielle pour réaliser le dépistage familial des sujets à risque. Références bibliographiques |
||||||||