La lettre de l’Hypophyse – Juillet 2014
 |
||||||||
|
||||||||
|
||||||||
 |
||||||||
|
Vous reprendrez bien une Newsletter avant l’été ? Madame, Monsieur, cher(e)s collègues, cher(e)s ami(e)s, « L’été s’ra chaud, l’été s’ra chaud dans les t-shirts, dans les maillots, D’la Côte d’Azur jusqu’à Rio… » Pour vos moments de détente, nous avons donc le plaisir de vous proposer cette nouvelle Newsletter Hypophyse dont le format pratique vous permettra de l’apporter sur la plage ou à un barbecue… Une Newsletter Hypophyse à déguster sans modération à l’apéritif ou à la mi-temps des matchs de la Coupe du Monde (pour les fans) ! Bonne lecture, bon été 2014 ! Frédéric Castinetti P.S. : Pour les prochaines Newsletters Hypophyse, les contributions du plus grand nombre d’entre vous sont souhaitées. Je ferai donc parvenir dès l’annonce de la date un mail vous proposant de contribuer à hauteur d’un résumé d’article ou d’une mise au point. Nous comptons sur votre aide. |
||||||||
 |
||||||||
|
LCI 699, un traitement anticortisolique prometteur D’après Bertagna X, Pivonello R, Fleseriu M et al. LCI699, a Potent 11?-hydroxylase Inhibitor, Normalizes Urinary Cortisol in Patients With Cushing’s Disease: Results From a Multicenter, Proof-of-Concept Study. J Clin Endocrinol Metab 2014;99(4):1375-83. Le LCI699 est un nouvel inhibiteur de la 11 ?-hydroxylase, développé par les laboratoires Novartis, dont la 1re étude « proof-of-concept » vient d’être rapportée dans le Journal of Clinical Endocrinology and Metabolism. Ce traitement, dont la demi-vie est de 4 heures, a aussi un effet inhibiteur de l’aldostérone synthase.
Quelle place pour le pasiréotide dans l’acromégalie ? Alors que les sous-types 2 (SSTR2) et 5 (SSTR5) des récepteurs de la somatostatine sont les plus représentés à la surface des cellules somatotropes, les analogues de la somatostatine (SSA) usuels, octréotide et lanréotide, se caractérisent par une affinité exclusivement dédiée à SSTR2. Le pasiréotide (ou SOM230), nouvel SSA multivalent fixant simultanément SSTR2 et surtout SSTR5, inhibe la secrétion de GH d’adénomes somatotropes humains in vitro mais son impact en clinique par rapport à l’octréotide demeurait inconnu. Quatre-vingt quatre centres, 27 pays, et 358 patients acromégales naïfs de tout traitement médical furent nécessaires pour conduire cette étude prospective pendant 12 mois (M12). Chaque patient fut randomisé, recevant mensuellement octréotide LP 20 mg (OCT+, n = 182) ou pasiréotide LP 40 mg (SOM+, n = 176). Une titration de la dose se justifiait à M3 et M7 s’il persistait une GH moyenne ? 2,5 µg/L et/ou une valeur d’IGF-1 ? LSN pour l’âge et le sexe. À M12, 31,3 % des patients du groupe SOM+ contre 19,2 % du groupe OCT+ (p = 0,007) attestaient d’un contrôle biochimique de la maladie (i.e. GH ? 2,5 µg/L ET valeur normale d’IGF-1 pour âge/sexe). Le traitement par pasiréotide ne s’accompagnait pas d’un effet antitumoral plus marqué à l’IRM. Les effets indésirables étaient similaires à l’exception d’une proportion plus importante de patients présentant une hyperglycémie (28,7 vs 8,3 %) ou un authentique diabète (19,1 vs 3,9 %) dans le groupe SOM+, effets indésirables identifiés du SOM230. L’étude s’est poursuivie depuis, avec la possibilité pour chaque patient soit (1) de rester dans leur bras de randomisation initiale (n = 120), soit (2) de basculer vers l’autre thérapie en cas de contrôle insuffisant (n = 119). Les résultats de (1) retrouvent une proportion équivalente de patients contrôlés à M25 dans les 2 groupes (48,6 % pour SOM+ et 45,7 % pour OCT+). Dans (2), leswitch opéré du pasiréotide LP vers l’octréotide LP n’augmentait pas le nombre de patients contrôlés alors que la substitution en sens inverse permettait un contrôle biochimique chez 17,3 % de patients supplémentaires à M25 (Posters SUN-88-95, Endocrine Society 2013). Une tendance récemment confirmée par les résultats de l’étude prospective multicentrique PAOLA. Cette dernière interessait des patients acromégales non-contrôlés de façon adéquate sous octréotide LP 30 mg ou lanréotide autogel (ATG) 120 mg, qui furent randomisés en 3 groupes comme suit : 1) SOM230 = 40mg LP/mois (n = 65) ; 2) SOM230 = 60 mg LP/mois (n = 65), ou 3) poursuite du traitement initial par octréotide LP ou lanréotide ATG (n = 68). Au terme des 24 semaines de traitement, 15,4 % et 20 % de patients des groupes 1 et 2 respectivement attestaient d’un contrôle biochimique sur la GH et l’IGF-1, contre aucun patient du groupe 3 (Poster P907, ECE 2014). En objectifs secondaires, une proportion supérieure de patient des groupes pasiréotide LP 40 et 60 mg ont présenté une diminution du volume tumoral > 25 % (18,5 % et 10,8 %) versus octréotide LP ou lanréotide ATG (1,5 %). Les signes et symptômes cliniques étaient généralement plus améliorés dans les groupes pasiréotide LP 40 et 60 mg, soulignant l’intérêt thérapeutique significatif du pasiréotide chez certains patients acromégales non contrôlés par les SSA actuellement disponibles.
Étude PRIMARYS : du neuf dans le traitement de 1re ligne de l’acromégalie ? Certaines études se sont intéressées aux effets des analogues somatostatinergiques comme traitement de 1re intention dans l’acromégalie, mais des lacunes méthodologiques persistent, compromettant la bonne évaluation de ces traitements. L’étude PRIMARYS est une étude prospective, multicentrique, ouverte et sans bras contrôle. Les critères d’inclusion étaient des patients acromégales, naïfs de tout traitement, avec une sécrétion de GH > 1 ng/mL après hyperglycémie provoquée par voie orale (HPO), une IGF1 supérieure à la normale pour le sexe et pour l’âge, un macro-adénome, et un champ visuel normal. Le critère de jugement principal était la proportion de patients pour qui nous avions une réduction du volume tumoral d’au moins 20 % à la 48e semaine de traitement. |
||||||||
 |
||||||||
|
Quel test génétique dans les adénomes hypophysaires ? Les progrès dans la chimie des réactions de séquence selon la méthode de Sanger et très prochainement, l’arrivée des nouvelles techniques de séquençage à haut et moyen débit dans le diagnostic de routine (type NGS – Next Generation Sequencing) rendent les analyses génétiques plus accessibles à la pratique clinique courante avec un rendu de résultats de plus en plus court. Néanmoins, il ne faut pas perdre de vue l’objectif de ces tests génétiques : améliorer la prise en charge du patient et donner un conseil génétique adapté. Une demande d’analyse génétique non adaptée peut aboutir à la découverte d’un variant allélique de signification inconnue pouvant inquiéter inutilement le patient et sa famille. Il est donc important de suivre les recommandations fondées sur les observations épidémiologiques. Les gènes impliqués dans les adénomes hypophysaires sont des gènes suppresseurs de tumeurs. Deux types d’anomalies moléculaires peuvent être à l’origine d’une inactivation du gène : des microlésions visibles par séquençage (mutation non-sens, faux sens, microdélétion et insertion à l’origine d’un décalage du cadre de lecture) et des macrolésions, non visibles par séquençage, telles que les délétions d’une partie plus ou moins importante du gène, appelées réarrangement de grande taille (RGT). Ces RGT, diagnostiqués en routine par la technique de MLPA (Multiples Ligation Probe Amplification) sont rares et représentent entre 1 et 5 % des anomalies moléculaires retrouvées sur ces gènes. Caractéristiques cliniques des adénomes hypophysaires dues à des mutations de NEM1 ou de AIP Les données observationnelles ont permis de préciser les spécificités du phénotype des adénomes hypophysaires dues à des mutations de NEM1 ou de AIP. Dans la NEM1, l’adénome hypophysaire est présent chez 46 % des patients (1). Il est la première manifestation de la maladie dans 21 % des cas et est isolé dans 15 % d’entre eux (Delemer B, unpublished data). Il s’agit dans 78 % des cas d’un prolactinome et dans 50 % d’un macro-adénome. Ce sont des tumeurs particulières par leur volume, leur agressivité (2) et leur résistance au traitement avec une absence normalisation de l’hypersécrétion dans 58 % des cas (1). Alors que l’adénome hypophysaire dû à une mutation NEM1 est fréquemment un prolactinome, l’adénome hypophysaire dû à une mutation de AIP est dans 80 % somatotrope (3). Il s’agit aussi d’une tumeur plus volumineuse et agressive, plus résistante au traitement (AIP étant un des maillons de la chaîne de transduction intracellulaire des analogues de la somatostatine) [4]. Dans 80 % des cas cette tumeur hypophysaire survient chez les patients avant l’âge de 30 ans, avec un dimorphisme sexuel (61 % chez les hommes). Alors que la pénétrance de la NEM1 liée à l’âge est de 100 % au-delà de 50 ans (5), la pénétrance de l’adénome hypophysaire en cas de mutation de AIP est incomplète et mal connue (30 à 60 %) [3]. Quelles analyses génétiques dans les adénomes hypophysaires familiaux ? L’hyperparathyroïdie est la première manifestation de NEM1 puisqu’elle est présente chez 98 % des patients (1). Ainsi, devant tout adénome hypophysaire, la présence d’une hyperparathyroïdie doit être écartée et l’établissement de l’arbre généalogique doit rechercher un antécédent familial d’adénome hypophysaire et d’hyperparathyroidie ou d’autres lésions « NEM1 » pour orienter l’analyse génétique. La présence d’un antécédent familial d’adénome hypophysaire sans aucun antécédent ni chez le patient ni chez ses apparentés de lésions « NEM1 », justifie une analyse du gène AIP complète (séquençage et recherche de RGT), quel que soit l’âge du patient. L’analyse du gène NEM1 est inutile. Mais en cas de doute sur une lésion « NEM1 » possible, chez le patient ou ses apparentés, une analyse du gène NEM1 pourra être associée, elle aussi complète (séquençage et recherche de RGT). Quelles analyses génétiques dans les adénomes hypophysaires sporadiques ? Pourquoi est-il légitime de poser la question d’une analyse génétique devant un tableau sporadique d’adénome hypophysaire : parce que 10 % des mutations de NEM1 sont des mutations de novo, parce que la pénétrance des mutations de AIP est incomplète. Il est clairement inutile de demander une analyse génétique devant tous les adénomes hypophysaires sporadiques, le pourcentage de mutation sur le gène AIP ne dépassant pas 3 %. À l’inverse, une demande d’analyse génétique de NEM1 et AIP est nécessaire chez les enfants (< 18 ans) devant tout adénome hypophysaire isolé sporadique quel que soit le phénotype. Le pourcentage de mutation du gène AIP peut atteindre 33 % (6) et 8 % pour la mutation de NEM1 (7). Le pourcentage de mutation de AIP est de 50 % en cas de gigantisme ! Au-delà de 18 ans, les données de littérature ont établi le seuil de l’âge à 30 ans en-deçà duquel une analyse génétique de NEM1 et AIP est recommandée, chez un patient porteur d’un adénome hypophysaire sporadique et isolé, quel que soit le type d’adénome mais seulement en cas de macroadénome. La fréquence des mutations de AIP ou NEM1 est d’environ 12 %. Une analyse par séquençage est suffisante, une recherche de RGT paraît inutile, ces dernières étant associées à une forte pénétrance de la maladie. Quel conseil génétique, quelle conduite à tenir chez les apparentés asymptomatiques dans le cadre des mutations de AIP ? Si le conseil génétique et la conduite à tenir chez les apparentés asymptomatiques sont clairement établis pour les mutations de NEM1, aucun consensus formel n’est encore publié pour les mutations de AIP. La pénétrance incomplète des mutations de AIP rend les choses complexes : il est inutile d’inquiéter un apparenté, porteur de la mutation familiale et qui ne développera peut être jamais un adénome hypophysaire. Ceci est particulièrement vrai pour les mutations de AIP découvertes dans un contexte d’adénome isolé sporadique. Parmi toutes les mutations de AIP mise en évidence dans ce contexte familial, seulement 2 adénomes hypophysaires chez 2 apparentés ont été rapportés dans la littérature, il s’agissait de micro-adénomes non fonctionnels. Néanmoins, voici quelques pistes, formulées à la lumière des règles établies pour les mutations de NEM1 et des études observationnelles sur les adénomes hypophysaires avec mutations de AIP. Ces adénomes survenant chez les gens jeunes et en particulier chez l’enfant, un dépistage génétique vers l’âge de 5 à 10 ans (comme pour le gène NEM1) paraît raisonnable. Les mutations de AIP pouvant être associées à tous les phénotypes hypophysaires, incluant les adénomes non fonctionnels, une IRM hypophysaire est requise lors du premier bilan. La fréquence de cette surveillance radiologique pourrait être tous les 3 à 5 ans comme pour le gène NEM1. Compte tenu de la fréquence des adénomes somatotropes, une surveillance de la courbe de croissance annuelle est justifiée chez l’enfant, plus ou moins associée à un dosage d’IGF1, PRL. Lorsque tout ce bilan est négatif, jusqu’à quel âge faut-il poursuivre cette surveillance ? Seulement 10 % des adénomes hypophysaires dus à des mutations de AIP ont été diagnostiqués après l’âge de 40 ans et 5 % après 50 ans (3), il n’est donc peut-être pas utile d’imposer une surveillance à vie. Lorsque le premier bilan est négatif, un arrêt de surveillance pourrait être proposé vers l’âge de 40 à 50 ans. Références bibliographiques
|
||||||||
 |
||||||||
|
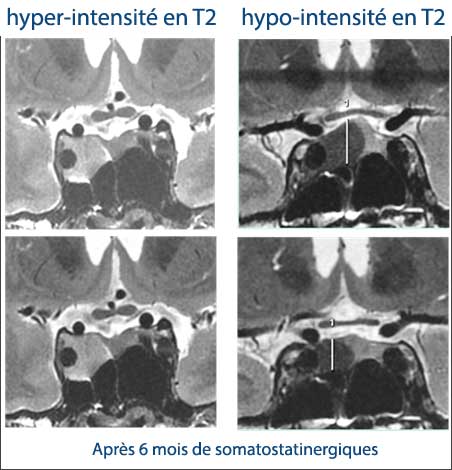
Intérêt du signal T2 des adénomes hypophysaires à GH, oui, mais quel est le tissu de référence ? Il est aujourd’hui admis que les adénomes hypophysaires somatotropes richement granulaires (densely granulated) ou présentant une cytokératine en périnucléaire (perinuclear pattern) répondent plus favorablement à un traitement par analogues de la somatostatine .
Références bibliographiques
|
||||||||