Hypothyroïdie congénitale : le « test de Guthrie », et après ?
Marie PUERTO, Bordeaux
D’après la communication SY-06 de Athanasia STOUPA (Paris)
Hypothyroïdie congénitale
Le dépistage néonatal à J3 de vie, basé sur le dosage de la TSH, permet aujourd’hui de dépister 300 bébés / an, soit environ 1 enfant sur 3000, atteints d’hypothyroïdie congénitale primaire (HCP). Ce dépistage est justifié par des signes cliniques modestes à la naissance, une relative fréquence de la maladie, un traitement simple, et un impact majeur puisque le devenir au long cours de ces patients sur les plans de la santé osseuse, du métabolisme et du développement cognitif, est similaire à celui de la population générale dès lors qu’ils sont dépistés précocement et pris en charge.
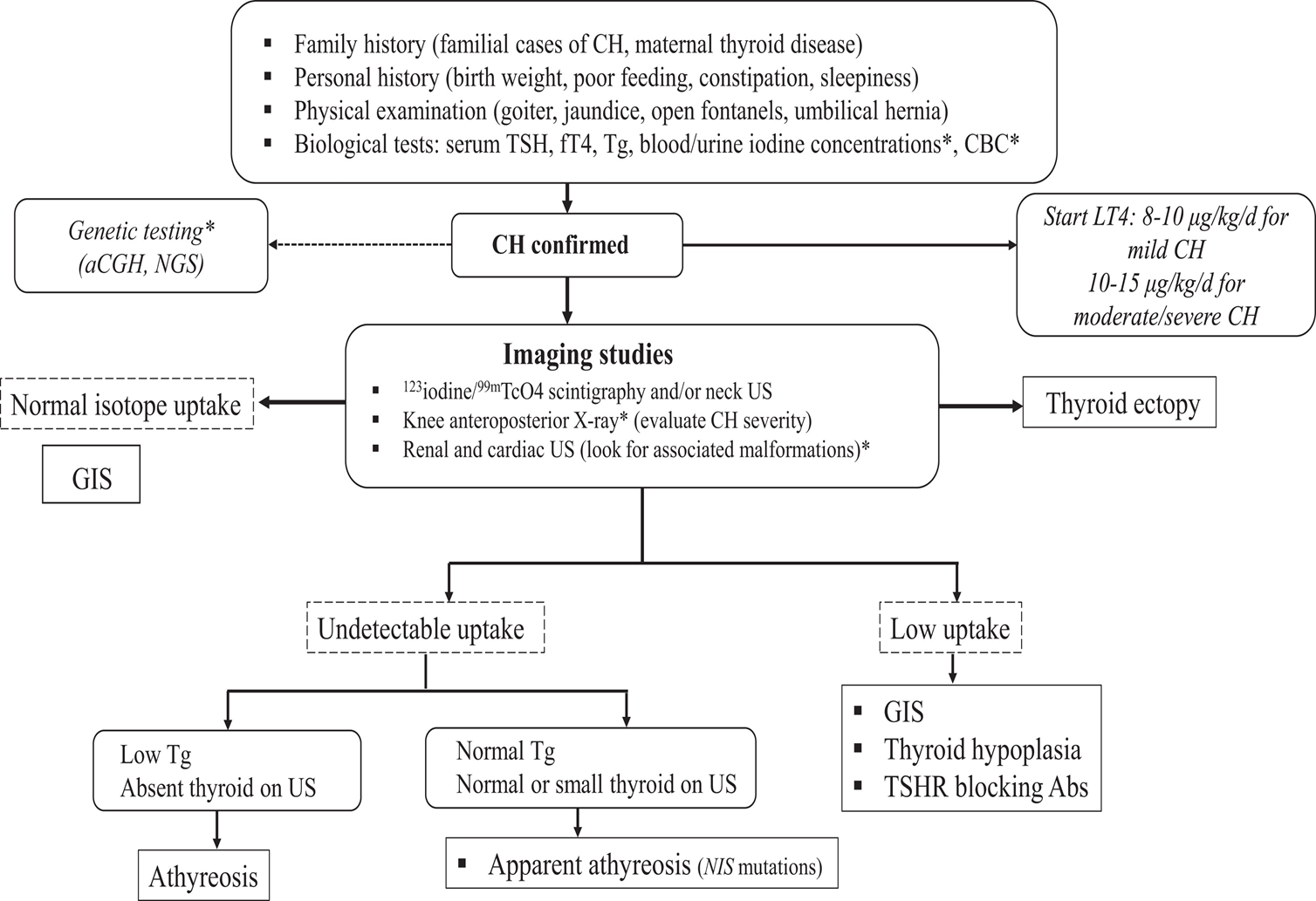
Les étiologies des HCP sont liées dans 65% des cas à des dysgénésies thyroïdiennes : athyréose, hypoplasie, hémi-thyroïde ou ectopie thyroïdienne. Dans 35% des cas, la thyroïde est bien en place, mais le patient présente des troubles de l’hormonosynthèse : ce sont les hypothyroïdies à glande in situ. Ces derniers patients peuvent présenter un goitre. La part d’hypothyroïdie avec glande in situ connait une augmentation nette sur les dernières années.
Le diagnostic étiologique est porté principalement par l’échographie et la scintigraphie (cf figure).
La génétique de l’hypothyroïdie congénitale est complexe, avec un taux de diagnostic génétique de l’ordre de 50% pour les hypothyroïdies à glande in situ, mais seulement de l’ordre de 5% pour les dygénésies thyroïdiennes.
Une hypothèse polygénique est retenue dans de nombreux cas, avec la « théorie des 2 hits » : 1er hit à composante héréditaire, en témoignent notamment des antécédents familiaux fréquents d’hypothyroïdie, et un 2ème hit sporadique, en témoigne la discordance de l’ordre de 92% sur l’HCP pour les jumeaux monozygotes.
Pour exemple, les mutations de PAX 8 et TTF1 peuvent être à l’origine aussi bien d’hypothyroïdie avec glandes in situ que d’hypoplasie voire d’athyréose thyroïdienne. Certaines mutations du gène FOX 1, lorsqu’elles sont bialléliques peuvent conduire au syndrome de Bamfort-Lazaruth avec hypothyroïdie congénitale et syndrome malformatif, mais les variations alléliques du gène FOX1 peuvent conférer une susceptibilité à l’hypothyroïdie congénitale. Les mutations de TSH-R qui confèrent des résistances à la TSH de sévérité variable, ont été décrites initialement comme de transmission récessive, mais dans certaines familles se manifestent par une transmission autosomique dominante.
Une indication d’analyse génétique est retenue devant toute hypothyroïdie congénitale présentant, ou bien des antécédents familiaux d’hypothyroïdie, ou bien un syndrome malformatif associé impliquant en priorité rein et cœur, ou bien en cas de consaguinité.
Les insuffisances thyréotropes, beaucoup plus rares (1/15 000 ou 20 000 enfants), ne sont pas identifiées par le dépistage néonatal. Elles se manifestent fréquemment en association avec d’autres déficits anté-hypophysaires qui font la symptomatologie principale.
Chez les patients adultes atteints d’HCP, ayant une thyroïde in situ et une dyshormonogenèse, une surveillance échographique est recommandée du fait d’un surrisque de nodule, de goitre et possiblement de cancers. Une surveillance de l’audition est également recommandée car les surdités sont plus fréquentes.
Stoupa et Al., JCEM 2022, Approach to the patient with congenital hypoparathyroidism