La lettre Surrénales de Juin 2015
|
Hypercortisolismes sévères, infertilité, tumeurs testiculaires, crises catécholaminergiques, diagnostic biologique…
|
||||||||
|
||||||||
|
Chers passionnés des surrénales, Nous avons le plaisir de vous présenter la 5e NewsLetter de la Surrénale ! Elle illustre une nouvelle fois la gravité et la diversité des pathologies surrénaliennes : hypercortisolismes sévères (J.B. Corcuff), infertilité, tumeurs testiculaires (J. Young et C. Bouvattier) et même de redoutables crises catécholaminergiques (O. Chabre). Heureusement ces articles vous apportent également des solutions, parfois très novatrices, pour traiter ou éviter ces complications redoutables. Sans oublier les progrès apportés au diagnostic biologique, pour qu’il reste la pierre angulaire de l’endocrinologie (V. Ducros) . Bonne lecture ! |
||||||||
 |
||||||||
|
Corticoïdes et crise catécholaminergique Il est bien connu que les patients porteurs d’un phéochromocytome (PH) ou d’un paragangliome (PGL) ont un risque élevé de crise catécholaminergique (CC). Celle-ci correspond à une décharge brutale de catécholamines, qui se manifeste par une hypertension artérielle (HTA) sévère, des troubles du rythme, une insuffisance cardiaque, un syndrome coronarien aigu.. qui peuvent être rapidement fatals. Ces crises, parfois révélatrices d’un PHPGL jusque là ignoré, sont déclenchées par différents facteurs : anesthésie (induction d’une anesthésie générale), chirurgie, pression abdominale, et certains médicaments. Il semble qu’il soit nécessaire de combler cette lacune, avec nuance, en effet plusieurs observations cliniques qui mettent en cause des corticoïdes exogènes dans le développement de CC ont été rapportées, y compris une observation très récente (1). Dans un article encore plus récent, Barrett fait une revue exhaustive de tous les cas recensés pendant 50 ans entre 1962 et 2013 (2). Ils retrouvent 25 cas de CC, qui se répartissent ainsi : – 3 sont liées à des tests de freinage fort (8 mg/j sur 2 jours) par la dexaméthasone, chez des patients explorés pour incidentalome surrénalien, dont une CC mortelle chez une jeune femme de 26 ans, dont les signes de CC sont apparus 36 heures après la première prise de dexaméthasone. Les 2 autres, dont les symptômes sont apparus 5 et 8 heures après la première prise de dexaméthasone ont survécu et tous les 3 avaient en commun d’avoir des phéochromocytomes, dont le plus grand diamètre était égal ou supérieur à 4 cm, avec densité radiologique sans injection > 28 UH. Il faut souligner qu’il n’y a pas eu de cas de CC rapportés avec des tests de freinage par 1 mg de dexaméthasone. – 16 CC sont liées à des traitements glucocorticoïdes, avec 3 décès. Ici les délais d’apparition de la CC étaient de 8 à 96 heures, et le type de corticoïdes et la voie d’administration sont très variables . Cinq patients avaient des symptômes évocateurs de PH avant le traitement glucocorticoïde, et les ATCD n’étaient pas renseignés pour 6 patients. Les PH faisaient de 2 à 6,8 cm. – Enfin, 1 CC rattachée à un test au synacthène synthétique (il s’agit de la cosyntropine, le produit actuellement disponible) 250 ug a été rapportée en 1990, chez une patiente qui recevait aussi une perfusion de dexaméthasone 1mg. Les symptômes sont apparus 5 minutes après l’injection de synacthène et ont disparu après 15 minutes, la patiente avait un PH de 5 x 7cm. À cela il faut ajouter 5 autres cas de CC après test à l’ACTH, dont 4 fatals, mais tous sont survenus avant 1970 avec des préparations animales d’ACTH. Les auteurs prennent le soin de préciser que leur étude ne permet pas d’affirmer un lien de cause à effet entre l’administration du corticoïde et la CC, mais en pratique que faut-il penser de ces observations inquiétantes ? Les auteurs prennent le soin de préciser que leur étude ne permet pas d’affirmer un lien de cause à effet entre l’administration du corticoïde et la CC, mais le doute est permis. Les auteurs proposent de :
|
||||||||
|
Hommes nés avec un bloc en 21 classique sévère : une enquête française montre que la prise en charge à l’âge adulte est loin d’être optimale et qu’il existe un risque important d’infertilité Le bloc enzymatique en 21-hydroxylase (B21) est de loin l’endocrinopathie génétique la plus fréquente (1). La forme dite classique (B21C) est cliniquement la plus sévère car elle s’accompagne déjà d’une insuffisance surrénale chez les 2 sexes pouvant entraîner une décompensation aiguë (2). Chez les patients de sexe féminin cette maladie, caractérisée par une hyperproduction de précurseurs androgéniques, est en plus responsable d’une masculinisation plus ou moins sévère des organes génitaux. Malgré les améliorations techniques des chirurgies génitales correctrices parfois itératives, ces femmes gardent des séquelles physiques et possiblement psychiques qui altèrent leur vie affective à l’âge adulte (2). En effet, la solitude est fréquente, la vie sexuelle souvent altérée, et les maternités assez rares (3). Se surajoutent des signes d’hyperandrogénie qui, associés à une taille diminuée, peuvent perturber l’image corporelle et l’estime de soi de ces femmes (3, 4). L’impact de ces séquelles sur la qualité de vie justifient la défense du traitement prénatal, mené avec courage par des équipes françaises (5) qui vise à prévenir la masculinisation génitale avant la naissance et donc empêcher son cortège délétère. Les travaux médicaux ayant évalué sur des séries significatives d’hommes adultes, la vie de couple, les fonctions gonadotropes, testiculaires et la fertilité sont quasiment inexistants (6). Un travail français, sous l’égide du Centre de référence des anomalies du développement sexuel, avec l’aide du Ministère de la santé (PHRC) et de l’association des patients « Surrénales » a donc été conduit, réunissant un grand nombre de CHU français (6). Il avait comme objectif d’évaluer une grande série d’hommes adultes nés avec un B21C. Cette étude a retrouvé certaines données comme la diminution de la taille finale. De façon intéressante, les mesures de la 17-hydroxyprogestérone, de l’ACTH et de la rénine, avec des valeurs souvent très élevées indiquaient que dans la « vraie vie » l’équilibre thérapeutique n’atteint que rarement les objectifs théoriques affichés par certains experts (2). Une découverte intéressante et inattendue a été une tendance modérée à l’hypotension. Elle pourrait être liée à une adhésion thérapeutique insuffisante et éventuellement à des apports diminués en sel. Une question intéressante est de savoir si cette diminution relative chronique de la pression artérielle pourrait protéger les hommes B21 d’accidents vasculaires. Mais la question d’une insuffisance surrénale infra-clinique chez ces patients mérite aussi d’être discutée, car elle pourrait précipiter la survenue de décompensations aiguës (7). L’évaluation hormonale de l’axe gonadotrope et des fonctions testiculaires a montré (6) des profils variables allant de 1) normalité (testostérone, LH, FSH normales : sujets eugonadiques) en passant par 2) des profils d’insuffisance en gonadotrophines hypophysaires (LH, FSH basses avec taux de testostérone variable) et 3) des tableaux évoquant une insuffisance testiculaire primitive (ITP) [FSH, LH élevées, testostérone variable et inhibine B basse]. L’analyse des échographies testiculaires faites chez 164 de ces hommes a montré volontiers une imagerie normale chez les sujets eugonadiques, une hypotrophie testiculaire dans les déficits gonadotropes et une forte prévalence de volumineuses tumeurs surrénaliennes intra-testiculaires (figure) [ les fameuses « inclusions » surrénales intra-testiculaires (ISIT) ] en cas de profil hormonal d’ITP. Les échographies testiculaires ont montré de plus la forte fréquence des ISIT qui a été décelée chez plus de 30 % de ces patients. Ces données suggèrent déjà que le dépistage des ISIT devrait être systématique au plus tard à partir de l’adolescence. Il permettrait de dépister ces tumeurs, à un stade infra-clinique, avant qu’une destruction du parenchyme testiculaire s’installe de façon irréversible et entraîne une ITP (figure). Une faille importante de la prise en charge des hommes B21C a été dévoilée par cette enquête : alors qu’il est établi depuis près de 15 ans que ces patients ont un risque important d’infertilité (8), seuls 71 (32 %) des 219 hommes suivis par des endocrinologues dans des CHU français avaient eu un spermogramme. En d’autres termes, près de 70 % de ces hommes adultes avec B21C n’avaient eu aucune évaluation spermatique. Cette réalité est d’autant plus inquiétante que lorsque l’analyse spermatique a été pratiquée, les spermogrammes montraient une azoospermie ou une oligospermie sévère dans 42 % des cas. Une amélioration des pratiques professionnelles semble donc nécessaire pour éviter que l’infertilité de ces malades se transforme en stérilité définitive. Pour atteindre ces objectifs la première étape serait de mieux diffuser, avec un esprit pédagogique, les travaux qui évaluent les conséquences et les complications de cette maladie auprès de toute notre communauté et en particulier auprès des jeunes endocrinologues. Pour préserver la fertilité des hommes B21C le message est donc simple : évaluation gonadique hormonale et échographique dès l’enfance, avec information parentale. À l’adolescence, éducation des patients et des parents sur le risque d’infertilité et, dès que possible, réalisation d’un spermogramme avec cryoconservation, ce qui permet d’envisager le futur avec sérénité. Surveillance échographique régulière des testicules pour améliorer le dépistage des « inclusions » et assurer une prise en charge précoce avant que l’ITP s’installe.
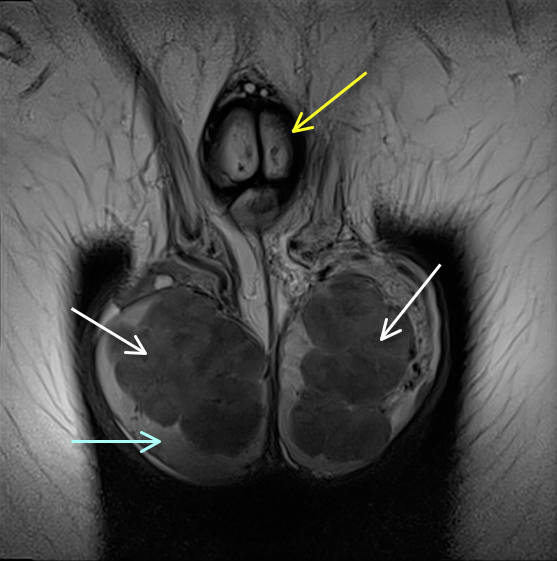
Figure. Aspect en IRM (coupe coronale en T1) d’inclusions surrénales intratesticulaires (ISIT) massives détruisant le parenchyme testiculaire chez un jeune homme de 24 ans atteint de forme classique de bloc en 21. Le spermogramme a montré une azoospermie. Malheureusement, chez ce patient, peu adhérent à la thérapeutique et au suivi, aucun dépistage échographique et aucune conservation de spermatozoïdes n’avaient été réalisés au cours de son suivi. Le pronostic de fertilité est très sombre. La flèche jaune montre le pénis en coupe coronale. Les flèches blanches les grosses tumeurs surrénales intratesticulaires et la flèche bleu clair le peu de parenchyme testiculaire restant (JY données non publiées). |
||||||||
|
Références bibliographiques 1. Coulm B, Coste J, Tardy V et al. DHCSF Study Group. Efficiency of neonatal screening for congenital adrenal hyperplasia due to 21-hydroxylase deficiency in children born in mainland France between 1996 and 2003. Arch Pediatr Adolesc Med 2012;166:113-20. 2. Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet 2005;365(9477):2125-36. 3. Gastaud F, Bouvattier C, Duranteau L et al. Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. J Clin Endocrinol Metab 2007;92(4):1391-6. 4. Merke DP, Poppas DP. Management of adolescents with congenital adrenal hyperplasia. Lancet Diabetes Endocrinol 2013;1(4):341-52. 5. Tardy-Guidollet V, Menassa R, Costa JM et al. New management strategy of pregnancies at risk of congenital adrenal hyperplasia using fetal sex determination in maternal serum: French cohort of 258 cases (2002–2011). J Clin Endocrinol Metab 2014;99(4):1180-8. 6. Bouvattier C, Esterle L, Renoud-Pierre P et al. Clinical outcome, hormonal status, gonadotrope axis and testicular function in 219 adult men born with classic 21-hydroxylase deficiency. A French national survey. J Clin Endocrinol Metab 2015;30:jc20144124. [Epub ahead of print] PMID: 25822101 7. Falhammar H, Frisén L, Norrby C et al. Increased mortality in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2014;99:E2715-21. 8. Cabrera MS, Vogiatzi MG, New MI. Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 2001;86(7):3070-8. |
||||||||
 |
||||||||
|
Vers une amélioration du dosage de l’aldostérone L’aldostérone, hormone stéroïdienne de poids moléculaire 360, est présente au niveau plasmatique à de très faibles concentrations (< 100 à 600 pmol/L). Jusqu’à ces dernières années, les seules méthodes de dosage disponibles étaient les méthodes d’immuno-analyses et plus particulièrement les méthodes par radio-immunoanalyses (RIA). Cependant, celles-ci présentent des inconvénients : un manque de spécificité des anticorps à l’origine de réactions croisées avec d’autres stéroïdes ou métabolites, des variations significatives des valeurs mesurées par des kits différents, enfin une influence de la dilution que l’on peut être amené à pratiquer lors de la mesure de l’aldostérone sur des prélèvements réalisés lors d’un cathétérisme des veines surrénaliennes, mais depuis une petite dizaine d’années, les techniques de chromatographie en phase liquide couplée à la spectrométrie de masse en tandem (LC-MS/MS), sensibles et plus spécifiques, se sont développées pour le dosage des stéroïdes et sont applicables au dosage de l’aldostérone. Qu’est-ce que la LC-MS/MS ? L’analyse par spectrométrie de masse en tandem repose sur l’obtention d’ions dits “pseudo-moléculaires” ou “ions-parents” ([M+H]+ en ionisation positive ou [M-H]- en ionisation négative) qui seront successivement triés, puis fragmentés en ions de plus petites masses mais caractéristiques de la structure de la molécule. Ces ions fragments, encore appelés ions-fils seront à leur tour triés et détectés. La spécificité de la mesure repose alors sur la filiation des fragments puisque l’on mesure une ou 2 transitions correspondant chacune à 1 ion-fils issus d’un ion-parent. La chromatographie en phase liquide qui est située en amont du spectromètre de masse permet de séparer les différents stéroïdes mais aussi les molécules de même masse (isobares) et ainsi de diminuer les interférences ; c’est une étape critique du couplage. Outre le gain en spécificité apporté par la mesure de transitions ions-parents/ions-fils, la LC-MS/MS apporte plus d’exactitude sur le résultat du dosage. En effet, comme l’on travaille sur la masse des molécules, on pourra ajouter en quantité connue lors de la préparation de l’échantillon, un étalon interne “marqué” c’est-à-dire la même molécule que la molécule à doser mais s’en différenciant par l’ajout d’isotopes stables comme le deutérium, le C13, ou l’N15. Cet étalon interne ayant le même comportement physico-chimique que la molécule à doser permettra de pallier les pertes lors de l’extraction, de la séparation chromatographique, de l’ionisation,… et in fine, d’améliorer le résultat. La quantification repose sur des gammes d’étalonnage réalisées en rapportant pour chaque point de gamme la surface du pic chromatographique du calibrateur à celle de l’étalon interne. Aldostérone et LC-MS/MS Outre sa concentration plasmatique en faible quantité, l’aldostérone est un composé polaire difficilement ionisable. De plus, à la différence de la plupart des autres stéroïdes, sa détermination est pratiquée en mode d’ionisation négatif, mode plus délicat à mettre en œuvre. Quoi qu’il en soit, ce dosage en LC-MS/MS est parfaitement réalisable en routine. L’étape de préparation de l’échantillon (plasma ou urine) est réalisée par extraction liquide-liquide ou solide-liquide à l’aide d’un solvant organique après ajout de l’étalon interne (aldostérone-D7). Cet extrait est concentré par évaporation sous azote puis injecté dans la colonne de chromatographie en phase liquide. Une bonne séparation chromatographique est nécessaire car d’autres stéroïdes co-extraits de masse très proche (exemple : cortisol, 18-hydroxycorticostérone de masse 362) pourront donner des fragments identiques et doivent donc être séparés de l’aldostérone pour ne pas apporter d’interférences. En travaillant en mode d’ionisation négatif, les transitions 359/189 pour l’aldostérone et 366/193 pour l’aldostérone-D7 sont suivies et quantifiées. Le rapport de surface des pics chromatographiques correspondant à ces 2 transitions sert à établir la gamme d’étalonnage. D’autres transitions sont également suivies comme 359/331 pour assurer la spécificité du dosage. La gamme de concentration est classiquement établie entre 30 et 3 500 pmol/L. Les coefficients de variations indiquant la précision du dosage sont inférieurs à 5-10 % en fonction des concentrations. La comparaison des résultats obtenus par LC-MS/MS à ceux des méthodes RIA montre le plus souvent une surestimation des résultats par RIA mais il existe des différences en fonction des trousses RIA. Un manque de standardisation des différents systèmes de dosage (RIA, immuno-essais automatisés, LC-MS/MS) est évident et doit être amélioré rapidement. Il n’existe pas encore ni de matériel de référence, ni de méthode de référence en LC-MS/MS pour le dosage de l’aldostérone. Autre point à souligner : la mise en place des méthodes par LC-MS/MS nécessite la révision des valeurs de référence et par là même, le choix des seuils pour le rapport aldostérone sur rénine. Les techniques par LC-MS/MS apportent un gain en spécificité non négligeable mais elles demandent un matériel coûteux qui ne peut être rentable que si l’on fonctionne en termes de plateforme pouvant réaliser plusieurs paramètres. Elles nécessitent aussi des mises au point qui peuvent être longues et un suivi continu des performances techniques. En conclusion, la LC-MS/MS apporte un gain réel sur la spécificité du dosage de l’aldostérone mais la standardisation des différentes méthodes de dosage plus adaptées à la routine comme les immuno-essais reste le point crucial toujours en attente ! Pour en savoir plus…
|
||||||||
|
Un traitement médical d’attaque pour les syndromes de Cushing graves Les syndromes de Cushing (SC) avec hypercortisolisme majeur, secondaires à un corticosurrénalome (CS) ou à un syndrome paranéoplasique (sPN), peuvent mettre en péril, à court terme, la survie des patients. Les principales causes de morbidité sont, en sus de la cachexie, de l’hypertension et de l’hypokaliémie, des infections sévères telles que celles à pneumocystis carinii ou des péritonites, des complications thromboemboliques telles que des embolies pulmonaires, etc. À ces facteurs pronostiques défavorables dus à l’histoire naturelle de la maladie s’ajouterait éventuellement le risque d’une chirurgie surrénalienne de sauvetage. L’usage de traitements médicaux rapidement efficaces serait d’un grand bénéfice dans ces formes graves. La métopirone et le kétoconazole sont des bloqueurs suffisamment rapides de la stéroïdogenèse pour que ces drogues soient évaluées en tant que traitement d’attaque de ces SC. Nous avons eu la possibilité d’évaluer rétrospectivement l’efficacité de la métopirone et du kétoconazole chez des patients avec un sPN ou un CS avec un hypercortisolisme intense (respectivement 14 et 8 patients). Ces syndromes graves occasionnaient hypertension (96 %), hypokaliémie (88 %) et diabète (75 %) avec des syndromes psychiatriques, des infections opportunistes, des phlébites et embolies pulmonaires, des fractures et/ou des escarres. Les bloqueurs de la stéroïdogenèse se sont montrés très efficaces. Après 1 semaine de traitement par métopirone et kétoconazole simultanément introduit chez la plupart des sujets, 50 % des sPN et 75 % des CS avaient une cortisolurie normalisée. Cette dernière continuait de décroître par la suite chez la grande majorité des patients. Après 1 mois de traitement, les prévalences de l’hypokaliémie, de l’hypertension et du diabète avaient réduit, parallèlement à une diminution de la nécessité de supplémentation potassique, d’antihypertenseurs et d’antidiabétiques. La tolérance a été satisfaisante : 3 patients (1sPN, 2 CS) ont arrêté le kétoconazole devant des transaminases hépatiques élevées (le patient avec sPN est décédé avec des métastases hépatiques 2 mois après l’initiation des traitements). Un peu moins de la moitié des patients se sont plaints de nausées modérées mais pour certains, du mitotane avait été introduit au cours du premier mois. Cette étude ainsi que d’autres avec des effectifs plus réduits montrent que ces 2 drogues sont rapidement efficaces sur plusieurs paramètres cliniques, métaboliques et hormonaux dans ces 2 étiologies de syndromes de Cushing graves. Les effets secondaires hépatiques surviennent dans environ 5 % des cas et leur apparition peut aisément être surveillée. Cette étude rétrospective ne permet pas de déterminer un effet-dose. Nous avons utilisé en moyenne 2,5 g/j de métopirone et 1 g/j de kétoconazole à l’initiation de traitement. Après quelques semaines de traitement, 2 attitudes seraient possibles : une diminution adaptative des doses ou une stratégie “block and replace” avec de l’hydrocortisone. La première diminuerait les prises de médications orales – souvent massives à l’initiation des traitements – permettant d’introduire du mitotane pour initier le traitement adrénolytique des CS. La seconde pourrait apporter plus de confort dans l’équilibre hormonal dans une optique de traitement symptomatique à moyen terme des sPN. La définition de la meilleure stratégie thérapeutique adaptée à chaque étiologie reste à préciser avec, de plus, l’arrivée possible de nouvelles molécules (kétoconazole non racémique, LCI699). Quelles que soient les molécules utilisées, il est important de noter que la surveillance biologique du blocage de la stéroïdogenèse repose sur la détermination de la cortisolurie ou éventuellement de la cortisolémie. Il sera donc très important d’utiliser des méthodes de dosage du cortisol qui ne présentent pas de réactions croisées notables avec les précurseurs stéroïdiens en amont du bloc causé par les 2 drogues et en particulier avec le 11-déoxycortisol. |
||||||||
|
Référence bibliographique Corcuff JB, Young J, Masquefa-Giraud P et al. Rapid control of severe neoplastic hypercortisolism with metyrapone and ketocorazole. Eur J Endocrinol 2015;172(4):473-81. |
||||||||
|
Infertilité des blocs en 21 classique chez l’homme : azoospermie avec grosses inclusions surrénaliennes intra-testiculaires traitée avec succès par mitotane, une approche médicamenteuse innovante mais… hors AMM Comme nous l’avons vu dans la lettre précédente, les hommes nés avec un bloc en 21 hydroxylase sévère « classique » ont un risque important d’infertilité par altération de la spermatogenèse (1). Les mécanismes responsables sont au minimum de 2 types : d’une part l’inhibition de la sécrétion des gonadotrophines hypophysaires LH et FSH, qui sont parfois effondrées (1), d’autre part la stimulation par l’ACTH de la croissance des inclusions surrénaliennes intra-testiculaires. Le premier mécanisme témoigne d’un hypogonadisme hypogonadotrophique (HH). Ce type d’altération de la spermatogenèse survient souvent chez les patients ayant arrêté ou diminué les thérapeutiques substitutives par hydrocortisone. L’augmentation de l’ACTH qui s’en suit est responsable de l’élévation majeure de PRG et 17OHP. Dans ces cas précis, une régression de l’azoospermie peut être obtenue par la réintroduction de l’hydrocortisone mais oblige parfois à freiner l’axe corticotrope par de la dexaméthasone (1,3). Cette approche est de temps en temps efficace mais induit après un certain délai un hypercorticisme inacceptable pour les patients. Il ne peut donc être utilisé au long cours. Le deuxième mécanisme : l’impact testiculaire des inclusions surrénaliennes intra-testiculaires (1,4) est directement lié à la stimulation par l’ACTH de ces reliquats corticosurrénaliens intratesticulaires, qui sont présents chez tous les hommes, mais ne font parler d’eux que chez les patients masculins B21C. Ces tumeurs peuvent empêcher directement l’excrétion de spermatozoïdes par 2 mécanismes indépendants, mais souvent intriqués : 1) d’une part, par une obstruction du hile testiculaire où convergent les tubes séminifères au niveau de rete-testis (4) ; il s’agit là d’une azoospermie par obstacle potentiellement réversible. L’autre mécanisme, irréversible celui-là, est tout simplement une destruction du parenchyme testiculaire et tout particulièrement des tubes séminifères (1,4). La littérature nous enseigne que le traitement chirurgical de ces cas est décevant avec parfois même une aggravation chirurgicale iatrogène des lésions testiculaires (4). Enfin, chez certains patients avec B21C, l’inhibition gonadotrope et l’atteinte testiculaire directe par les inclusions se conjuguent pour aggraver l’infertilité (1). La prise en charge devient alors très difficile et le traitement freinateur par la dexaméthasone est souvent inefficace. Dans ce contexte, il faut rappeler que ces inclusions tumorales sont composées de tissu corticosurrénalien. Ceci a été largement documenté aussi bien par les analyses biochimiques et moléculaires de ces tissus tumoraux (5) que par l’exploration hormonale et l’imagerie « in vivo » des patients (6, 7). À partir de ces notions nous avions pensé que l’utilisation d’un composé « adrénolytique » comme le mitotane pouvait être utile. Le but était double : détruire électivement le tissu tumoral d’origine surrénal au niveau testiculaire par une sorte de « thérapeutique ciblée » et profiter de l’effet antisécrétoire du mitotane pour empêcher la sécrétion des précurseurs stéroïdiens. Nous avons pu récemment valider cette approche thérapeutique chez un homme né avec un bloc 21C et une infertilité par azoospermie (8). Il présentait une double atteinte, un déficit en gonadotrophines par accumulation de PRG et 17OHP et des grosses inclusions testiculaires. L’administration de mitotane a entrainé 2 effets bénéfiques intéressants (figure), d’une part un effondrement de PRG et 17OHP qui a permis la levée de l’inhibition gonadotrope. D’autre part, une diminution importante du volume des inclusions par son effet antitumoral. Le plus spectaculaire a été la réapparition des spermatozoïdes dans l’éjaculat alors que de nombreux spermogrammes faits avant cette thérapeutique, y compris sous déxaméthasone, montraient l’absence de tout spermatozoïde. Une fécondation in vitro a pu être réalisée et un enfant en bonne santé est né (8). Au plan de la tolérance, une tendance à la majoration de l’insuffisance surrénale a été observée, très probablement secondaire au métabolisme accéléré de l’hydrocortisone et de la 9a-Fludrocortisone, car cet effet secondaire a régressé avec la majoration très importante des doses de ces 2 médicaments. Au plan hépatique il n’y a pas eu d’effet en dehors de l’élévation habituelle des gamma-GT sous mitotane. Ainsi, chez les hommes avec B21C azoospermes avec inclusions surrénaliennes intratesticulaires une ouverture thérapeutique existe maintenant. Pour conclure il faut souligner que le mitotane n’a pas d’autorisation de mise sur le marché dans cette indication. Sa prescription engage donc la responsabilité du médecin et ne doit être initiée que chez des patients parfaitement fiables après information loyale sur les bénéfices et les risques. Ce type de thérapeutique ne doit, d’autre part, être initiée que par des endocrinologues connaissant très bien le maniement de cette molécule, en milieu hospitalier et après une éducation thérapeutique approfondie pour prévenir les risques, en particulier celui d’insuffisance surrénale aiguë.
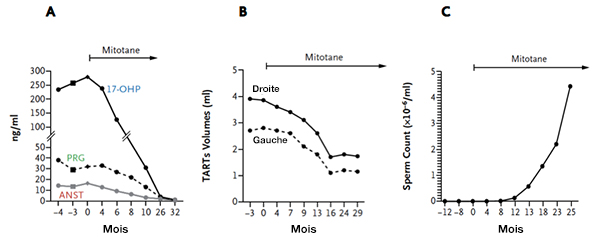
Figure. Homme né avec un bloc en 21 classique, présentant à l’âge adulte des grosses inclusions surrénaliennes intratesticulaires. Il consulte à l’âge de 29 ans pour une infertilité avec azoospermie. Un traitement par dexaméthasone n’a pas d’effet sur le volume des inclusions et l’azoospermie persiste. Un traitement par mitotane (3 g/j) a permis : A : une diminution de la 17-hydroxyprogesterone (17OHP), de la progestérone (PRG) et l’androstènedione (ANST) d’origine surrénalienne. B : une diminution spectaculaire du volume des inclusions et surtout, C : l’apparition de spermatozoïdes dans l’éjaculat. Une fécondation in vitro a été possible avec naissance d’un enfant en bonne santé, porteur hétérozygote de la mutation paternelle (adapté de la référence 8). |
||||||||
|
Références bibliographiques 1. Bouvattier C, Esterle L, Renoud-Pierre P et al. Clinical outcome, hormonal status, gonadotrope axis and testicular function in 219 adult men born with classic 21-hydroxylase deficiency. A French national survey. J Clin Endocrinol Metab 2015;30:jc20144124. [Epub ahead of print] 2. Tiitinen A, Välimäki M. Primary infertility in 45-year-old man with untreated 21-hydroxylase deficiency: successful outcome with glucocorticoid therapy. J Clin Endocrinol Metab 2002;87(6):2442-5. 3. Claahsen-van der Grinten HL, Otten BJ, Sweep FC, Hermus AR. Repeated successful induction of fertility after replacing hydrocortisone with dexamethasone in a patient with congenital adrenal hyperplasia and testicular adrenal rest tumors. Fertil Steril 2007;88:705.e5-8. 4. Claahsen-van der Grinten HL, Otten BJ, Stikkelbroeck MM, et al. Testicular adrenal rest tumours in congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab 2009;23:209-20. 5. Claahsen-van der Grinten HL, Otten BJ, Sweep FC et al. Testicular tumors in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency show functional features of adrenocortical tissue. J Clin Endocrinol Metab 2007;92(9):3674-80. 6. Young J, Couzinet B, Pholsena M et al. Plasma 3 beta-hydroxy-delta 5-steroids in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 1994;78(2):299-304. 7. Bernard V, Chougnet CN, Tenenbaum F, Young J. 131I-noriodocholesterol uptake by testicular adrenal rest tumors in patient with classical 21-hydroxylase deficiency. J Clin Endocrinol Metab 2014;99(11):3956-7. 8. Bry-Gauillard H, Cartes A, Young J. Mitotane for 21-hydroxylase deficiency in an infertile man. N Engl J Med 2014;371(21):2042-4. |
||||||||