|
VIEILLE CONNAISSANCE ET PARFAIT INCONNU : QUAND L’AMP CYCLIQUE RENCONTRE LE TATOU
Olivier Chabre (Grenoble)
Vieille connaissance : en descendant la voie jusqu’à PRKACA
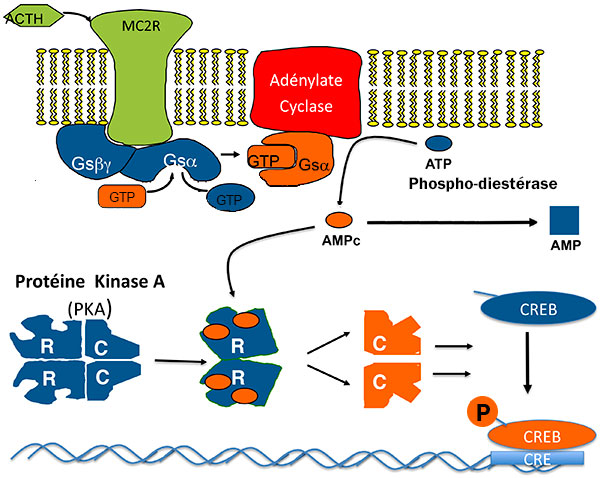
Pour stimuler la sécrétion et la prolifération de la cellule cortico-surrénalienne, l’ACTH active les maillons de la voie de l’AMP cyclique : ACTH, récepteur de l’ACTH (MC2R), Gs, adénylate cyclase, PKA et enfin les multiples cibles de PKA dont CREB (Cyclic AMP responsive Element Binding protein), facteur de transcription qui se lie à une séquence d’ADN “CRE” (figure 1).
Figure 1. Voie de l’AMPC dans la cellule corticosurrénalienne : cibles de la tumorigénèse.

AMP : adénosine monophosphate ; ATCH : adrénocorticotrophine ; CRE : Cyclic AMP responsive Element ; CREB : Cyclic AMP responsive Element Binding protein ; GDP : guanosine diphosphate : GTP : guanosine triphosphate.
Il était très tentant de rechercher une activation anormale d’un de ces maillons dans les tumeurs ou hyperplasies cortico-surrénaliennes responsables de syndrome de Cushing ACTH-indépendant. C’est ce qui a été fait avec succès ces 20 dernières années, qui ont vu l’identification de plusieurs mutations ou surexpressions. La plupart augmente le taux d’AMPc : expression anormale d’un récepteur couplé à Gs, très rares mutations activatrices du MC2R ou de Gs?, mutations inhibitrices de la phosphodiestérase qui hydrolysent l’AMPc, (donc activation par double négation !), nettement plus fréquentes.
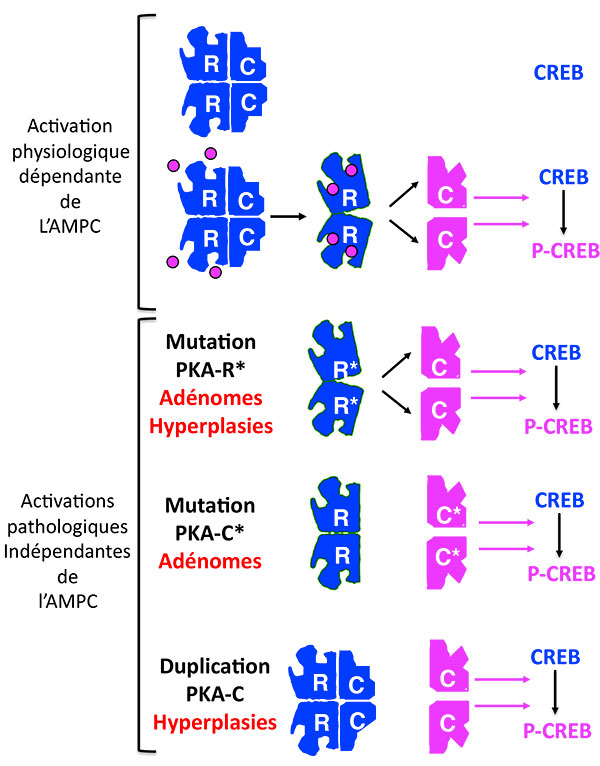
Au-delà de l’AMP cyclique, surgit la PKA dont il faut rappeler le fonctionnement : en l’absence d’AMPc, elle est un bloc inactif de 4 unités, les 2 sous-unités régulatrices bâillonnant les 2 sous-unités catalytiques. En présence d’AMPc, le bâillon saute et les sous-unités catalytiques phosphorylent leurs substrats. Les mutations déjà décrites de la PKA inhibent les sous-unités régulatrices, qui ne sont plus capables de lier les sous-unités catalytiques, même en l’absence d’AMP cyclique (figures 1 et 2).
Figure 2. Les activations multiples de la Protéine Kinase A.
PKA : protéine Kinase A ; CREB : Cyclic AMP responsive Element Binding protein ; P-CREB : CREB phosphorylé.
Le dernier né des mécanismes touche directement la sous-unité catalytique, qui répond à l’acronyme scatologique “PRKACA”, et dont l’activation a été rapportée dans un remarquable article qui a uni les équipes allemande, américaine et française (1), puis dans 3 autres articles américains, chinois et japonais (2-4). Cette activation est en fait la plus fréquente de toutes les activations de la voie AMPc et elle se fait de 2 façons différentes. Sous forme de mutation ponctuelle somatique dominante, présente à l’état hétérozygote seulement dans les cellules tumorales, elle est responsable selon les équipes respectives, de 37 % (22/59) [1], 52 % (34/65) [4] et jusqu’à 65 % (57/87) [3] des adénomes responsables de syndrome de Cushing. Il s’agit alors presque toujours d’une mutation du codon 617 qui touche la Leucine 206, acide aminé qui joue un rôle important dans la liaison de la sous-unité catalytique avec la sous-unité régulatrice.
Sous forme de duplication germinale, présente dans toutes les cellules, l’activation est liée à la saturation des sous-unités régulatrices par un excès de sous-unités catalytiques, et elle touche 14 % (5/35) [1] des hyperplasies corticosurrénalienne bilatérales, aussi responsables d’un syndrome de Cushing (figure 2).
Au total, les vieilles connaissances ont toujours un dernier tour dans leur sac… mais est-ce bien le dernier ?
Vieille connaissance : au départ était l’ACTH
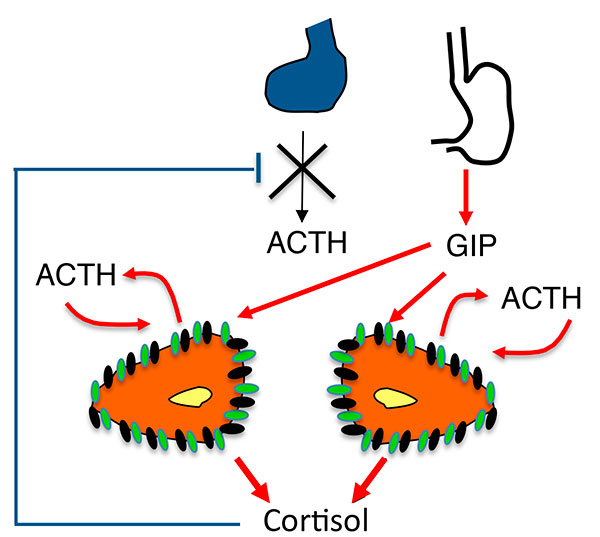
Les travaux sur la PRKACA cités plus haut apportent des données nouvelles et passionnantes, mais les techniques utilisées de séquençage à haut débit ont un caractère “rouleau compresseur” sans originalité. Tout autre est la nature, et donc le mérite, des Rouennais (5) qui par des techniques autrement élégantes arrivent à convaincre le lecteur que pour activer la voie de l’AMP cyclique rien ne vaut l’ACTH elle-même, produite sur place par des ilôts de cellules contenus dans les hyperplasies macronodulaires bilatérales.
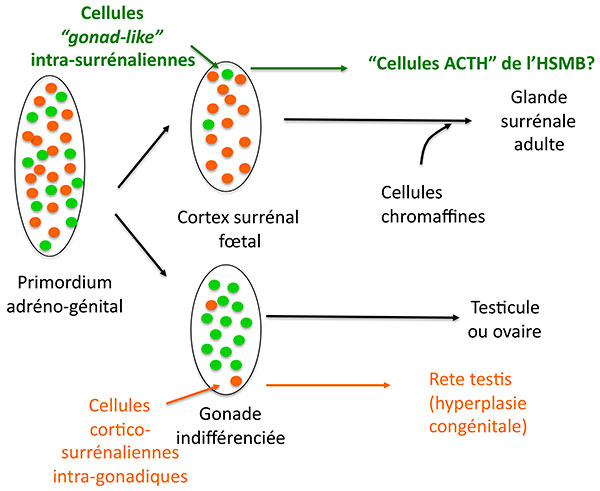
Ces cellules ont leurs particularités : si elles expriment plusieurs protéines caractéristiques des cellules stéroidogéniques comme SF1, 17 hydroxylase, le récepteur HDL, elles n’expriment pas l’antigène MHC de classe II normalement exprimé dans les cellules cortico-surrénaliennes, mais expriment en revanche un marqueur des cellules de Leydig et lutéales : insulin like 3. L’hypothèse séduisante des auteurs est qu’il s’agit de cellules originaires du primordium adréno genital (figure 3), qui se retrouvent dans la corticosurrénale de façon tout à fait symétrique aux ilôts cortico-surrénaliens présents dans les testicules, (qui sont à l’origine des “rete testis” dans l’hyperplasie congénitale). Il faut noter que ces cellules, qui savent cliver la POMC, ne fonctionnnent cependant pas comme des cellules hypophysaires : pas de stimulation de leur sécrétion d’ACTH par le CRH ni de freinage par la dexamethasone.
Figure 3. Hypothèse sur l’origine des cellules surrénaliennes sécrétant de l’ACTH au cours du développement du cortex surrénal.
ATCH : adrénocorticotrophine.
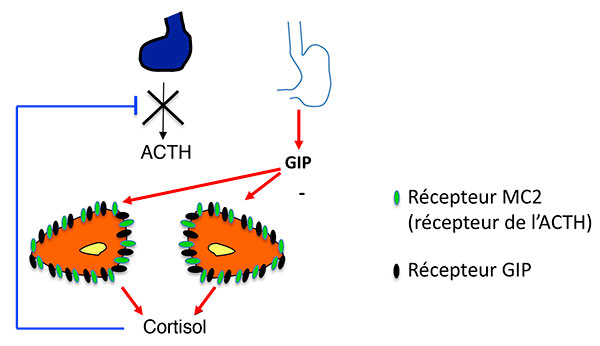
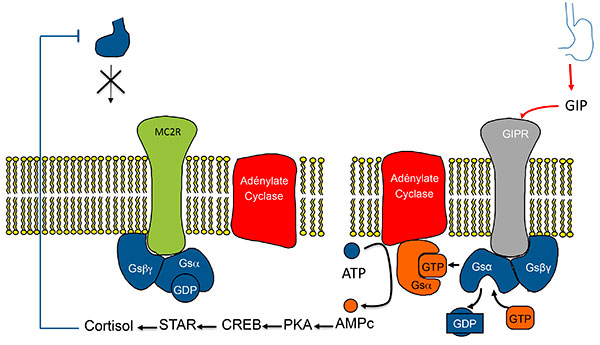
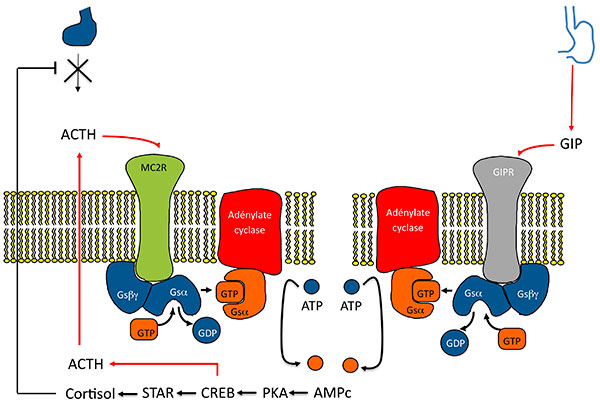
Quel lien entre cette production surrénalienne d’ACTH (véritable cauchemar pédagogique pour les enseignants en endocrinologie) et l’activation des récepteurs ectopiques, autre trait des hyperplasies macronodulaires bilatérales ? Il est complexe et Estelle Louiset et Hervé Lefebvre mettent en évidence un mécanisme qui enrichit le modèle des récepteurs ectopiques : ceux-ci n’agiraient pas seulement en stimulant eux-même l’AMP cyclique (figures 4A et B), mais également en stimulant la production d’ACTH, qui à son tour renforce la production d’AMP cyclique (figures 5A et B).
Figure 4.
A. Stimulation de la sécrétion de cortisol par expression ectopique de RCPG dans les HSMB. Modèle classique (exemple du GIPR).
ATCH : adrénocorticotrophine ; GIP : Gastric inhibitory Peptide ; GIPR Gastric inhibitory Peptide Receptor ; HSMB : hyperplasies surrénaliennes macronodulaires bilatérales.
B. Stimulation de la sécrétion de cortisol dans les HSMB. Activation de la voie de l’AMPC dans le modèle classique.
Activation de la voie de l’AMPc par expression anormale d’un RCPG (ici le GIPR). Modèle 1 : l’activation du RCPG “remplace” l’activation du récepteur de l’ACTH.
AMPC : adénosine monophosphate cyclique ; CREB : Cyclic AMP responsive Element Binding protein ; GDP : Guanosine diphosphate ; GIP : Gastric inhibitory Peptide ; GIPR Gastric inhibitory Peptide Receptor ; GTP : guanosine triphosphate ; HSMB : hyperplasies surrénaliennes macronodulaires bilatérales ; PKA : protéine Kinase K ; STAR : Steroid Acute regulatory Protein.
Figure 5.
A. Stimulation de la sécrétion de cortisol dans les HSMB. Nouveau modèle : sécrétion surrénalienne d’ACTH.
ATCH : adrénocorticotrophine ; GIP : Gastric inhibitory Peptide ; HSMB : hyperplasies surrénaliennes macronodulaires bilatérales.
B. Mécanismes de la sécrétion surrénalienne d’ACTH dans les HSMB.
Activation de la voie de l’AMPc par expression anormale d’un RCPG . Modèle 2 : l’activation du RCPG (ici GIPR) entraîne indirectement une activation du récepteur de l’ACTH par stimulation d’une sécrétion paracrine D’ACTH.
AMPC : adénosine monophosphate cyclique ; CREB : Cyclic AMP responsive Element Binding protein ; GDP : guanosine diphosphate ; GIP : Gastric inhibitory Peptide ; GIPR Gastric inhibitory Peptide Receptor ; GTP : guanosine triphosphate ; HSMB : hyperplasies surrénaliennes macronodulaires bilatérales ; PKA : protéine Kinase K ; STAR : Steroid Acute regulatory Protein.
Très bien me direz-vous, mais la voie de l’AMP cyclique peut-elle tout expliquer dans ces hyperplasies surrénaliennes macronodulaires bilatérales (HSMB) ? En effet, celles-ci posent un défi à l’endocrinologue : il y a une disproportion évidente entre leur taille, qui est monstrueuse – avec des masses qui atteignent 20 fois celles de surrénales normales –, et leur taux de secrétion qui est ridicule, souvent à peine supérieur à celui de surrénales normales, 20 fois plus petites. Comment tirer au clair ce paradoxe ? C’est ici qu’intervient un nouvel acteur, découvert lui aussi par une équipe française.
Le parfait inconnu : ARMC5
Armadillo est le nom espagnol du tatou, petit mammifère d’Amérique latine qui porte sur son dos une véritable armature. Le gène ARMC5 code pour une protéine qui contient des “armadillo repeat domains”, structures répétitives qui permettent de créer un domaine rigide de la protéine (un peu comme la carapace du tatou), au sein duquel elle peut interagir avec de multiples partenaires, pour assurer différentes fonctions. La caténine, protéine qui joue un rôle important dans la tumorigénèse corticosurrénalienne (voie Wnt) contient elle aussi des “armadillo repeat domains”.
Que vient faire ce tatou dans la corticosurrénale ? C’est bien évidemment aussi des techniques d’analyses moléculaires systématiques qui ont permis de débusquer l’inconnu : G. Assie et al (6) sont partis de l’hypothèse puissante qu’une pathologie bilatérale des corticossurénales avait toutes les chances d’être génétique, comme cela avait été retrouvé dans un petit nombre de familles.
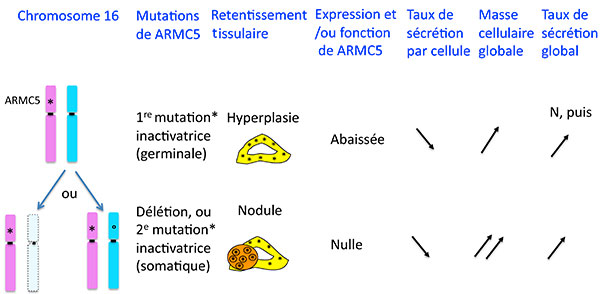
En comparant l’ADN tumoral et génomique de patients porteurs d’HSMB, ils ont identifié une région du chromosome 16 qui avait des délétions fréquentes, pour finir par identifier un gène dans cette région qui était muté sur un allèle de l’ADN génomique des patients et délété ou muté sur l’autre allèle dans les multiples nodules surrénaliens des patients. Ceci suggérait que ARMC5 se comporte comme un gène suppresseur de tumeur : la perte de fonction d’un allèle serait responsable d’une hyperplasie et la perte d’un autre allèle d’une tumeur (figure 6). En accord avec cette hypothèse, les auteurs montrent dans un modèle cellulaire que l’inhibition de l’expression de ARMC5 inhibe l’apoptose, ce qui a pour effet d’augmenter la population cellulaire.
Figure 6. Mutations de ARMC5 dans l’hyperplasie surrénalienne macronodulaire bilatérale.
ARMC5 est-il alors la vraie explication de tout ce qui se passe dans les hyperplasies macronodulaires bilatérales et en particulier rend-il compte de leur hypersecrétion ACTH “indépendante” ? Il y a ici un paradoxe apparent : G. Assie et al démontrent, toujours dans un modèle cellulaire, que l’inhibition d’ARMC5 diminue l’expression des enzymes de la stéroidogènèse nécessaires à la secrétion de cortisol : comment une mutation qui a pour effet de diminuer la stéroidogénèse peut-elle être responsable d’une pathologie caractérisée par une hypersécrétion de cortisol chez le patient ? L’explication logique est que les mutations perte de fonction ou délétion de ARMC5 ont un effet d’augmentation de la masse corticosurrénalienne qui au fil du temps, arrive à surcompenser la baisse du taux de secrétion de chaque cellule, et on conçoit que lorsqu’on arrive à une masse 20 fois plus importante, des cellules qui secréteraient 10 fois moins de cortisol qu’une cellule normale arriveront ensemble à secréter 2 fois plus qu’une glande normale (figure 6 ).
Très bien, mais à cet “effet masse”, on peut objecter qu’une cellule normale elle, est capable d’arrêter complètement sa secrétion de cortisol lorsqu’il n’y a plus de sécrétion hypophysaire d’ACTH : pourquoi les cellules ARMC5- n’en feraient-elles pas autant ?
C’est ici qu’il faut sortir à nouveau la carte “activation de l’AMP cyclique” : l’activation de récepteurs ectopiques et la sécrétion surrénalienne d’ACTH sont sans doute les facteurs qui permettent aux glandes géantes ARMC5 – géantes handicapées de la stéroïdogénèse –, de continuer à secréter un peu de cortisol, alors que l’hypophyse dit “Stop” (figure 7).
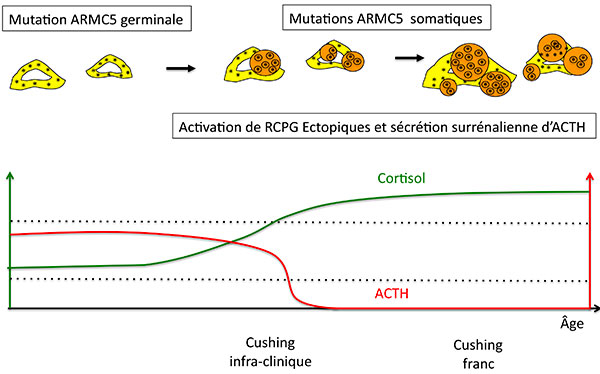
Figure 7. Progression de l’hyperplasie nodulaire et de la sécrétion de cortisol chez les patients porteurs de mutation germinale de ARMC5.
ACTH : adrénocorticotrophine ; RCPG : récepteur couplé aux protéines G.
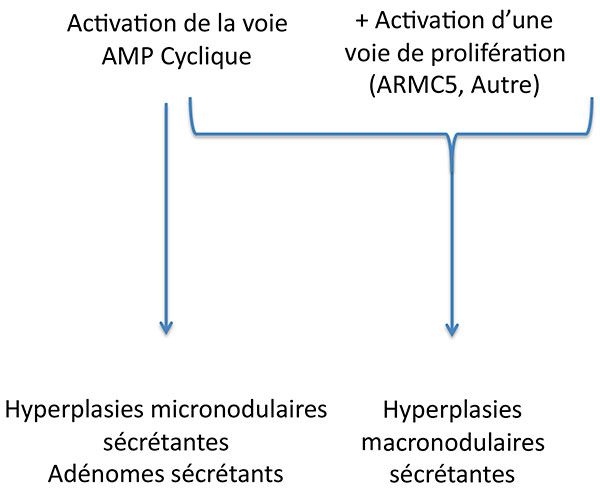
Au total, le syndrome de Cushing surrénalien de ces patients est sans doute le fruit du mariage improbable du tatou avec l’AMPc (figure 8) .
Figure 8. Complémentarité des voies d’activation dans la tumorigénèse corticosurrénalienne.
AMPC : adénosine monophosphate cyclique.
L’histoire s’arrête-t-elle là ? Sans doute pas : beaucoup de travail reste à faire pour mieux comprendre ARMC5, qui par ailleurs n’est retrouvé “que” dans un peu plus de la moitié des patients porteurs d’HSMB (6, 7) et, en outre, d’autres joueurs s’invitent déjà dans la danse – DOT1L et HDAC9 (3), des histones methyltransférase et déacétylase – qui suggèrent que des modifications de structure de la chromatine ont aussi leur mot à dire… à suivre !
|