
| Accédez au site |
 |
Lettre N°5 - Juin 2015 |
[ÉDITORIAL] [L'ACTUALITÉ COMMENTÉE] • La macroprolactinémie revisitée - |
• Intérêt des mesures répétées du cortisol salivaire pour le dépistage précoce des récidives post-chirurgicales de la maladie de Cushing - Antoine Tabarin, Bordeaux [MISE AU POINT SUR…] [IMAGES COMMENTÉES] |

La recherche française hypophysaire se porte bien
Frédéric Castinetti, CHU La Timone, Marseille
La recherche clinique hypophysaire française et francophone se porte bien… Aussi, quand il a fallu réfléchir aux brèves qui constitueraient cette newsletter, il a été facile de contacter directement les auteurs francophones de ces publications. Je me doute que certains parmi vous me taxeront de chauvinisme exacerbé, d’optimisme béat, de naïveté touchante, etc. Et pourtant… 6 études cliniques publiées dans les très bons journaux de la spécialité sur une thématique hypophysaire entre fin 2014 et début 2015, avec des résultats ayant un impact sur notre pratique quotidienne... Quand on considère la difficulté croissante de publier dans ces revues, on ne peut que se féliciter !
Vous aurez donc droit à :
- un bon tiers de prolactine - macroprolactinémie revisitée (Lyon), prolactinome de l’enfant et l’adolescent (Bicêtre),
- un grand tiers d’hypercorticisme - cortisol salivaire (Bordeaux), test couplé DexMinirin (Marseille), IRM des adénomes corticotropes silencieux (Foch-Ambroise Paré),
- un succulent tiers d’acromégalo-gigantisme, avec une mise au point sur GPR101 par le plus français des Belges (Pr A. Beckers),
- et un petit tiers avec l’image commentée du désormais plus belge des Français (Pr JF Bonneville)
Avouez que ça donne envie…
Bonne lecture, bonnes vacances et à très bientôt pour une nouvelle newsletter !

Risque de récidive post-opératoire dans la maladie de Cushing, quel test pour le suivi ?
Pauline Le Marc'hadour, Marseille
D’après Le Marc’hadour P., Castinetti F. et al. Postoperative follow-up of Cushing’s disease using cortisol, desmopressin and coupled dexamethasone-desmopressin tests: a head-to-head comparison.
Définir le plus précocement possible le devenir des patients opérés pour une maladie de Cushing reste à l’heure actuelle un challenge important afin d’adapter au mieux la surveillance au risque de récidive et proposer un suivi individualisé. L’objectif de cette étude était d’évaluer la valeur du cortisol plasmatique en post-opératoire immédiat, du test au Minirin® (desmopressine) et du test au Dex-Minirin® (test couplé dexaméthasone-desmopressine) comme prédicteurs du devenir en post-opératoire.
Il s’agissait d’une étude rétrospective, bicentrique (Marseille et Grenoble), qui a inclus 67 patients opérés par chirurgie transsphénoïdale pour une maladie de Cushing, en rémission en post-opératoire immédiat et avec un suivi minimal de 18 mois. Tous les patients avaient eu un dosage du cortisol plasmatique en post-opératoire immédiat, puis avaient été suivis à 3-6 mois en post-opératoire, et annuellement, avec un dosage du CLU (cortisol libre urinaire) sur 24 heures (2 à 3 mesures), un cycle nycthéméral de l’ACTH (adrénocorticotrophine) et du cortisol plasmatique, un test de freinage minute (1 mg de dexaméthasone), un test au Minirin® puis un test au Dex-Minirin®. Des courbes ROC avaient été utilisées pour définir les valeurs optimales du taux de cortisol plasmatique en post-opératoire immédiat, et de l’ACTH et du cortisol après test au Minirin® et au Dex-Minirin® à 3-6 mois en post-opératoire, comme facteurs prédictifs précoces de récidive en comparaison aux marqueurs classiques de récidive. Les tests au Minirin® et au Dex-Minirin® avaient ensuite été évalués comme marqueurs du devenir au cours du suivi. Au total, 11 patients avaient récidivé après une médiane de suivi de 52 mois (18-180). Le cortisol plasmatique en post-opératoire immédiat restait le “Gold standard” comme facteur prédictif précoce de rémission, une valeur inférieure à 35 nmol/L permettant de prédire une absence de récidive avec une valeur prédictive négative (VPN) de 93 %. Au cours du suivi, le test au Dex-Minirin® était plus précis que celui au Minirin®, et permettait de prédire l’absence de récidive avec une VPN de 100 % dans les 3 premières années de suivi. Il était particulièrement intéressant parmi les patients qui avaient eu un cortisol plasmatique supérieur à 35 nmol/L en post-opératoire immédiat, permettant de sélectionner 56 % de ces patients à faible risque de récidive lorsqu’il restait négatif. La positivité du test au Dex-Minirin® était définie à partir des courbes ROC par une augmentation de l’ACTH et du cortisol plasmatique supérieur à 50 % par rapport aux valeurs basales. Ce test était reproductible, avec une réponse concordante au fil des évaluations chez 91 % des patients. Les valeurs prédictives positives de chacun des marqueurs étaient décevantes, autant en post-opératoire qu’au cours du suivi. Au total, associer le test au Dex-Minirin® au cours des 3 premières années de suivi au cortisol en post-opératoire immédiat permettrait un suivi plus optimal des patients opérés pour une maladie de Cushing, en sélectionnant un sous-groupe de patients à faible risque de récidive malgré un cortisol en post-opératoire non optimal, et pour lesquels la surveillance pourrait être espacée. Cependant, aucun des tests ne permettait de prédire avec certitude le risque de récidive en cas de valeur positive, et un suivi rapproché devrait être poursuivi chez tous ces patients.
La macroprolactinémie revisitée
Lucile Parlant-Pinet, Lyon
D'après Parlant-Pinet L, Harthé C, Roucher F et al., Macroprolactinaemia: a biological diagnostic strategy from the study of 222 patients. Eur J Endocrinol. 2015 [Epub ahead of print]
La chromatographie par filtration sur gel est la référence pour le diagnostic de macroprolactinémie (MPRL), mais c’est une technique manuelle, longue et coûteuse. Afin de limiter le nombre de chromatographies nécessaires, nous avons évalué 2 méthodes de dépistage de la MPRL : 1) la récupération de la prolactine (PRL) après précipitation au polyéthylène glycol (PEG) par dosage RIA : récupération = PRL dans le surnageant après précipitation/PRL dans le sérum natif ; 2) le ratio des résultats de PRL issus de 2 techniques de dosage ayant des réactivités croisées différentes vis-à-vis des formes de PRL de haut poids moléculaire : ratio = PRL par RIA (forte réactivité croisée)/PRL par ECLIA sur automate Cobas e 601 (faible réactivité croisée).
Nous avons étudié 222 sérums, sélectionnés aléatoirement parmi les 386 sérums hyperprolactinémiques faisant l’objet d’une prescription de recherche de MPRL, reçus sur une période de 18 mois. Une MPRL a été diagnostiquée chez 64 patients, définie par une proportion majoritaire de big-big-PRL ou de big-PRL, d’après le profil d’élution issu de la chromatographie. L’ensemble des sérums avec une récupération inférieure à 30 % et/ou un ratio supérieur à 1,83 (22,1 % des sérums) présentaient un profil de MPRL à la chromatographie. À l’inverse, la chromatographie montrait une prédominance de PRL monomère pour l’ensemble des sérums avec une récupération supérieure à 80 % et/ou un ratio inférieur à 0,64 (33,3 % des sérums), ainsi que pour tous les sérums avec une récupération supérieure à 65 % et un ratio inférieur à 1 (48,6 % des sérums). La chromatographie était nécessaire pour affirmer ou exclure une MPRL lorsqu’aucun de ces critères n’était rempli, soit pour 27,9 % des sérums. La récupération après PEG est une méthode largement utilisée pour le diagnostic de MPRL. Dans notre étude, elle présentait une “zone grise” s’étendant de 30 à 80 %, qui comprenait 49,1 % des sérums. La concentration de PRL monomère peut être calculée comme suit, d’après les résultats de la chromatographie : PRL monomère = (% de PRL monomère/100) x PRL par RIA. En cas de MPRL, la PRL par ECLIA (de réactivité croisée faible mais non nulle) et la PRL dans le surnageant après précipitation au PEG n’étaient que faiblement corrélées à la concentration de PRL monomère. Ces 2 approches ne permettaient donc pas une estimation fiable de la concentration de PRL monomère en cas de MPRL. L’association de 2 tests de “dépistage” en préalable permet de s’affranchir de 72,9 % des chromatographies sur gel avec une sensibilité de 100 % pour le diagnostic des formes MPRL.
Macroprolactinomes chez l'enfant et l'adolescent : bonne réponse au traitement agoniste dopaminergique
Philippe Chanson, Kremlin-Bicêtre
D'après Salenave S., Ancelle D., Bahougne T. et al. Macroprolactinoma in children and adolescents : factors associated with the response to treatment in 77 patients. J Clin Endocrinol Metab 2015;100(3):1177-86.
Les adénomes hypophysaires sont rares chez l’enfant et l’adolescent. La moitié des patients à cet âge ont un adénome à prolactine. Dans certains cas une mutation, en particulier du gène AIP (23 % des enfants de la série d’adénomes de Bicêtre) ou du gène de la ménine, est à l’origine de ces adénomes.
Grâce à une analyse rétrospective d’une cohorte de jeunes patients présentant un macroprolactinome (adénome à prolactine de plus de 10 mm de plus grand diamètre), menée aux CHU de Bicêtre, de Reims et de Lyon, on dispose maintenant d’un peu plus d’éléments sur la présentation clinique et la réponse au traitement des macroprolactinomes survenant dans cette population.
Les patients âgés de moins de 20 ans au moment du diagnostic de macroprolactinome et vus dans l’un de ces 3 centres entre 1983 et 2013 ont été étudiés et leur profil génétique caractérisé. La réponse hormonale et tumorale aux agonistes dopaminergiques a été analysée et cela après un suivi moyen de 8,2 ± 5,8 années.
La cohorte comportait 77 patients (26 garçons et 51 filles) dont l’âge moyen au diagnostic était de 16,1 ± 2,5 ans (entre 4,5 ans et 20 ans). Dix-huit avaient moins de 15 ans au diagnostic et une toute petite minorité (2 garçons) avait moins de 10 ans. Dans les 2 sexes, le symptôme révélateur le plus fréquent était un trouble pubertaire à type de retard pubertaire ou d’aménorrhée primaire ou secondaire (49 %), suivi par des troubles visuels en rapport avec un macroprolactinome comprimant le chiasma optique (24 %). Un retard de croissance était observé dans 24 % des cas. On peut aussi noter une prise de poids dans 24 % des cas : d’ailleurs au moment du diagnostic la moitié des enfants étaient obèses ou en surpoids. La prolactinémie basale était significativement supérieure chez les garçons en comparaison des filles (7 168 ng/ml chez les garçons versus 1 433 ng/ml chez les filles). Le volume des adénomes était également plus important avec un diamètre maximum de 33 ± 14 mm chez les garçons versus 19 ± 9 mm chez les filles. Ils étaient aussi plus souvent invasifs chez les garçons.
Une mutation du gène AIP été trouvée chez 5 des 55 patients testés (9 %) et de celui de la ménine chez 3 des 59 patients testés (5 %).
Ces macroadénomes à prolactine ont bien répondu au traitement par agonistes dopaminergiques puisque, dans 74 % des cas, la prolactinémie s’est normalisée sous traitement. Quatorze des 20 patients qui n’ont pas totalement répondu (c’est-à-dire qui n’ont pas normalisé leur prolactinémie) aux agonistes dopaminergiques avaient aussi une réponse insuffisante de l’adénome aux agonistes dopaminergiques mais globalement le traitement par agoniste dopaminergique a permis une réduction parfois spectaculaire du volume de l’adénome chez 76 % des patients.
Les patients qui ne normalisaient pas leur prolactinémie sous agoniste dopaminergique avaient en général une valeur plus élevée et un volume tumoral supérieur, les 2 paramètres étant bien sûr étroitement liés. Enfin, la présence d’une mutation de l’un ou l’autre des 2 gènes était un facteur prédictif indépendant de la résistance aux agonistes dopaminergiques.
En conclusion, les adénomes à prolactine sont rares avant 20 ans et surviennent plutôt chez les filles et à l’adolescence. Leur phénotype est assez comparable à celui des adultes : les tumeurs sont plus grosses et plus invasives chez les garçons, chez les adolescentes, il s’agit plus souvent de microadénomes. Enfin, leur réponse aux agonistes dopaminergiques est excellente, justifiant, comme chez l’adulte, leur prescription en première intention… même en cas de volumineuse lésion comprimant le chiasma optique.
Intérêt des mesures répétées du cortisol salivaire pour le dépistage précoce des récidives post-chirurgicales de la maladie de Cushing
Antoine Tabarin, Bordeaux
D'après Danet-Lamasou M, Asselineau J, Perez P et al. Accuracy of repeated measurements of late-night salivary cortisol to screen for early-stage recurrence of Cushing's disease following pituitary surgery. Clin Endocrinol (Oxf). 2015 Feb;82(2):260-6.
L’histoire naturelle des récidives de maladie de Cushing est mal documentée dans la littérature, mais l’expérience commune atteste que celles-ci s’installent souvent de manière progressive avec une ambiguïté clinique et biologique lors des stades précoces de la récidive. Le cortisol salivaire vespéral qui permet d’apprécier la rupture du rythme nycthéméral de sécrétion du cortisol a fait la preuve de son utilité pour le diagnostic du syndrome de Cushing patent. Pourrait-il être un marqueur précoce de la récidive ? Cette question était l’objet d’une étude menée par l’équipe d’endocrinologie du CHU de Bordeaux. Au-delà des performances diagnostiques et du fait de la variabilité importante de la sécrétion de cortisol dans le syndrome de Cushing, les auteurs ont également cherché à déterminer le nombre de prélèvements de cortisol salivaire vespéral utiles pour dépister la récidive à un stade précoce.
Cette étude rétrospective a concerné 36 patients opérés pour maladie de Cushing au CHU de Bordeaux et qui étaient considérés comme en rémission en période postopératoire immédiate. Dans le cadre du suivi, ces patients ont eu de multiples prélèvements de cortisol salivaire vespéraux faits au domicile. Les résultats de ceux-ci ont été comparés à un bilan biologique exhaustif contemporain réalisé en milieu hospitalier incluant cortisolurie des 24 heures, dosage du cortisol à minuit, test à la desmopressine, test de freinage à la dexaméthasone. Les patients ont été considérés en rémission lorsque l'ensemble des tests hospitaliers apparaissaient normaux, ou à un stade précoce de la récidive lorsque 2 d'entre eux étaient anormaux. Pour parler de stade précoce de la récidive, le cortisol libre urinaire devait être ou normal ou très modestement élevé (inférieur à 2 fois la limite supérieure de la normale). La réalité de la récidive a été confirmée chez la majorité des patients avec la progression clinique et biologique de la maladie dans le suivi. Les valeurs du cortisol salivaire ont été comparées entre les patients considérés en rémission et en récidive précoce. Le nombre de dosages de cortisol salivaire utiles a été déterminé à travers une modélisation mathématique particulière.
Les conclusions des auteurs sont que le cortisol libre urinaire est effectivement un marqueur tardif de la récidive de la maladie de Cushing puisque celui-ci est normal chez 61 % des patients qui sont en récidive précoce. Le cortisol salivaire apparaît en revanche être un marqueur sensible pour dépister la récidive précoce. Cependant, il existe une très grande variabilité entre les valeurs de cortisol salivaire à un stade de récidive précoce qui alternent entre normales et pathologiques. Dans cette situation, les auteurs remarquent que les performances optimales (sensibilité et spécificité à 90 %) sont obtenues lorsqu’on considère les valeurs de 3 ou 4 cortisols salivaires vespéraux.
Le cortisol salivaire réalisé en ambulatoire peut donc être une investigation utile au dépistage du syndrome de Cushing et être aussi performant qu’une série de tests complexes devant être réalisés en milieu hospitalier. Il est toutefois nécessaire d'effectuer 3 à 4 prélèvements salivaires à des jours distincts pour des performances de dépistage optimales. Les données de cette étude rétrospective seront à confirmer afin de pouvoir être recommandées en pratique.

Comment la réflexion clinique peut-elle aboutir à une innovation génétique :
l’exemple des Géants
Frédéric Castinetti, CHU La Timone, Marseille
Dans son numéro du 3 décembre 2014, le New England Journal of Medicine publiait un article sur un nouveau syndrome appelé X-LAG (X-linked Acro-Gigantism). Cet article était publié conjointement par le service d’endocrinologie du CHU de Liège dirigé par le Professeur Albert Beckers et par l’Unité d’endocrinologie pédiatrique du NIH dirigée par le Docteur Constantine Stratakis. Il y a une quinzaine d’années, le Professeur Beckers décrivait pour la première fois une nouvelle entité familiale hypophysaire qu’il a dénommée FIPA pour “Familial Isolated Pituitary Adenomas”. Les caractéristiques phénotypiques de ce nouveau syndrome ont été publiées en 2006 (1). En 2007, l’équipe liégeoise montrait que 15 % des patients FIPA ont une mutation du gène AIP (2). Les adénomes mutés pour AIP sont le plus souvent des adénomes à GH (Growth hormone). En 2010, la même équipe montrait que les adénomes AIP positifs surviennent beaucoup plus tôt (souvent avant 20 ans) et sont beaucoup plus agressifs que les adénomes somatotropes AIP négatifs (3). Dans le même article, les auteurs faisaient remarquer que le gigantisme était beaucoup plus fréquemment rencontré dans les adénomes AIP positifs, ce qui était logique étant donné l’âge d’apparition de la maladie. Cette observation a conduit l’équipe liégeoise à lancer une grande étude internationale sur le gigantisme et 3 ans plus tard, les données cliniques, biologiques, génétiques de 200 géants étaient collectées à travers le monde (4).
Parmi les centres participatifs, figurait celui du NIH. Dix géants issus de ce centre faisaient partie de la série et de façon intéressante, dans 2 situations (l'une sporadique et l’autre familiale), une microduplication au niveau du chromosome X avait été mise en évidence par cGH. Cette microduplication comportait 13 gènes et aucun n’était connu pour être impliqué dans la régulation de l’hormone de croissance. Malgré des recherches intenses, aucun autre cas du NIH n’avait pu montrer les mêmes caractéristiques génétiques. C’est alors que les 2 chercheurs ont décidé d’unir leurs efforts. D’un côté, il y avait une microduplication dont la signification était inconnue et de l’autre une grande collection d’ADN de géants. C’est alors que sur la base des renseignements fournis par le Docteur Stratakis, l’équipe liégeoise a entrepris les études de cGH sur les ADN et très rapidement, la réponse apparaissait évidente. En même temps que se réduisait le nombre de gènes impliqués dans la microduplication, apparaissait très clairement un nouveau syndrome, que les auteurs ont appelé X-LAG et qui présente les caractéristiques montrées ci-dessous. Les enfants naissent tous normaux mais très rapidement, toujours avant 3 ans et très fréquemment avant 1 an, quelquefois même dès l’âge de 3 mois, ils commencent à grandir de façon effrénée. Lorsque le diagnostic d’une croissance anormale est réalisé, l’examen IRM de la région hypophysaire met en évidence des signes d’hyperplasie ou, si le patient est étudié un peu plus tard, un adénome volumineux. Il s’agit presque toujours d’un adénome mixte secrétant des quantités réellement phénoménales d’hormone de croissance et de prolactine dans la plupart des cas. Si les patients ne sont pas traités, ils ont tout le loisir de grandir pendant une très longue période et c’est ce qui explique qu’ils peuvent atteindre des tailles assez gigantesques comme l’a fait dans le passé le plus grand homme du monde, Robert Wadlow, 2,72 mètres, ou, dans le présent, Sultan Kosin, 2,51 m. Au stade actuel, 4 gènes sont dupliqués dans tous les cas. La duplication de 2 des gènes impliqués intervient dans d’autres syndromes et ne conduit pas au gigantisme. Restaient 2 gènes dont l’un apparaît surexprimé dans les tumeurs hypophysaires de ces patients comme l’ont montré les auteurs. Il s’agit de GPR101. Celui-là constitue donc le coupable idéal pour expliquer ce nouveau syndrome. GPR101 est un récepteur couplé à une protéine G, exprimé physiologiquement dans le noyau arqué de l’hypothalamus et dans l’hypophyse. Le ligand est à ce jour inconnu. L’étude de plus de 200 patients acromégales au niveau somatique montre dans 4 % des cas une modification (E308D) qui est également rencontrée dans la population normale mais dans des proportions bien moindres. Il pourrait s’agir d’une mutation activatrice. Les études de transfection dans des cellules GH3 montrent que cette modification entraîne une augmentation significative de sécrétion d’hormone de croissance par rapport au récepteur sauvage. X-LAG est un nouveau syndrome qui peut apparaitre de façon sporadique ou familiale. Deux familles sont décrites. Le nombre de patients familiaux est petit mais au stade actuel, le mode de transmission paraît dominant et la pénétrance apparaît complète. Il s’agit donc aussi du deuxième gène impliqué dans le FIPA. En résumé, voici un nouveau syndrome phénotypiquement bien caractérisé et qui explique les plus grandes tailles jamais observées. Un nouveau mécanisme génétique d’apparition d’adénome hypophysaire est montré, une nouvelle voie impliquée dans la croissance est mise en évidence et une nouvelle cause de FIPA est définie. Cet article porte un éclairage nouveau sur la pathologie hypophysaire. Il génère de très nombreuses questions sur les mécanismes impliqués et offre potentiellement des perspectives thérapeutiques nouvelles.
Références bibliographiques
1.Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab 2006;91(12):4769-75.
2. Daly AF, Vanbellinghen JF, Khoo SK et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab 2007;92(5):1891-6.
3. Daly AF, Tichomirowa MA, Petrossians P et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab. 2010;95(11):E373-83.
4. Rostomyan et al. ENDO 2013

Les adénomes corticotropes silencieux
Laure Cazabat, CHU Ambroise Paré
D'après Cazabat L., Martin Dupuy M., Boulin A. et al. Silent but not unseen : multi-microcystic aspect on T2-weighted MRI in Silent Corticotroph Adenomas Clinical endocrinology 2014;81(4):566-72.
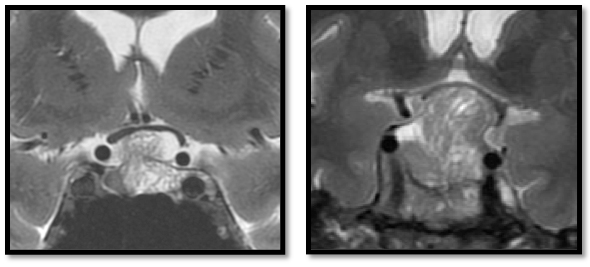 Adénome corticotrope silencieux : aspect multi-microkystique en T2
Adénome corticotrope silencieux : aspect multi-microkystique en T2
Les adénomes corticotropes silencieux (Silent corticotroph adenomas, SCA) sont des macroadénomes découverts sur un syndrome tumoral, souvent des troubles visuels, et diagnostiqués à l’examen anatomopathologique avec une immunohistochimie positive pour l’ACTH.
Ils se présentent donc comme des macroadénomes non fonctionnels. Ils sont dits “silencieux” sur l’absence d’hypercorticisme. Cependant, chez la plupart des patients, en l’absence de signes cliniques évidents, une recherche d’hypersécrétion corticotrope n’est pas réalisée.
Le Dr Anne Boulin a noté un aspect particulier de ces adénomes corticotropes “silencieux” à l’IRM sur la séquence pondérée en T2. L’objectif de l’étude était d’estimer la valeur prédictive pour le diagnostic de SCA de cet aspect particulier à l’IRM : la présence de multiples microkystes sur les séquences T2.
Si les SCA peuvent être suspectés à l’imagerie, la recherche d’un hypercortisolisme modéré ou infraclinique pourra être ciblée et ainsi en limiter les coûts.
Nous avons étudié les IRM préopératoires de 17 patients avec SCA et les avons comparés à l'IRM préopératoire de 2 groupes : 14 macroadénomes corticotropes sécrétants (MCS) et 35 macroadénomes non fonctionnels gonadotropes (MNFG). Le choix de ces 2 groupes permet la comparaison avec la présentation clinique habituelle (MGNF) et avec les MCS afin de rechercher un continuum entre MCS et ACS.
Tous les patients ont été opérés à l'hôpital Foch. Toutes les IRM ont été relues par 2 médecins et par la neuroradiologue en aveugle. Les aspects IRM sont classés en multi-microkystique et absence de microkyste.
À noter que les 4 autres SCA sont des macrokystes (4/17).
Dans notre série sur 1 096 patients opérés, 312 sont des MNFG et 29 des SCA ; les SCA représentent 8,5 % des non-fonctionnels. En extrapolant les résultats de notre étude à la cohorte entière, on peut estimer que devant un macroadénome non-fonctionnel, s'il existe un aspect multi-microkystique en T2 (12 % des cas), c’est un SCA dans 55 % des cas, sinon c’est un MNFG dans 98 % des cas.
Au total, devant un macroadénome hypophysaire non fonctionnel, la présence de multiples microkystes en T2 à l’IRM est un très bon outil pour le diagnostic de SCA avant la chirurgie. Nous proposons d’étudier finement l’axe corticotrope devant tout patient avec un adénome non fonctionnel ayant de multiples microkystes en T2.
Cette notion pourrait être ajoutée à la conférence de consensus français de 2012 sur la prise en charge des macroadénomes non fonctionnels en cours de rédaction.
Une hypophyse “convexe” chez une adolescente
J.-F. Bonneville*, F. Cattin**
* Département d’endocrinologie (Pr A.Beckers), CHU de Liège
** Service de neuroradiologie, CHU de Besançon
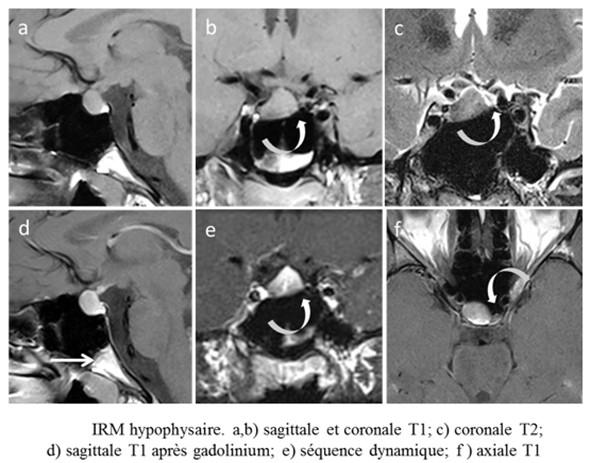
La découverte fortuite au scanner ou en IRM d’une “grosse” hypophyse chez une adolescente est fréquente et interroge : certes, l’hyperplasie hypophysaire physiologique de la puberté est évoquée, mais le clinicien ne retiendra en général ce diagnostic qu’après un bilan biologique répété, une surveillance étroite y compris par IRM… mais parfois un doute persiste. L’arrière-pensée d’une tumeur hypophysaire – adénome ou kyste de la poche de Rathke –, d’une hypothyroidie primaire voire d’une hypophysite est souvent présente. L’analyse de la morphologie sellaire et du sinus sphénoïdal peut ici aider. Ainsi, une petite selle turcique est responsable d’une inadaptation contenu/contenant : l’hypophyse, bien que de volume normal, donne l’illusion de sortir anormalement de la selle, à la manière d’une brioche débordant de son moule. Les causes d’une étroitesse de la selle turcique sont multiples : constitutionnelles, dorsum sellae épais, épine sellaire… mais pas toujours évidentes pour un œil non exercé. Plus facile à détecter – à condition d’y penser –, est l’hyperpneumatisation du sinus sphénoïdal ; il existe en effet fréquemment une corrélation entre grand sinus sphénoïdal et petite selle turcique, comme si l’expansion du sinus venait s’opposer à la formation de la selle. L’observation suivante illustre cette variante anatomique trompeuse. Marion, 16 ans, diabétique, fait une chute avec perte de connaissance à la suite d’un malaise hypoglycémique. La découverte au scanner d’une “grosse” hypophyse entraîne la réalisation d’une IRM dont le compte-rendu inquiète le médecin et les parents : comment les rassurer ? Certes, l’hypophyse semble haute et est proche du chiasma optique ; mais le signal est normal et homogène en T1 (a,b) et en T2 (c) ; de même le rehaussement après injection de gadolinium (d ) ; de même une séquence dynamique (e), réalisée dans un second temps. Mais c’est l’analyse du sinus sphénoïdal qui emporte la conviction : sa pneumatisation s’étend en arrière jusqu’à la synchondrose sphéno-occipitale (flèche en d ), ce qui est inhabituel chez une enfant de 16 ans ; cette hyperpneumatisation, asymétrique, soulève et refoule l’hypophyse (flèches courbes) ; le plancher sellaire est non pas abaissé à droite mais surélevé à gauche. Le diagnostic est donc ici celui de variante anatomique chez une adolescente et aucun suivi ne sera nécessaire.
