
| Accédez au site |
 |
Lettre N°2 - Juin 2012 |
[ÉDITORIAL] Du neuf dans la prise en charge médicamenteuse de la maladie de Cushing + À consulter ici : la fiche Anticortisoliques et maladie de Cushing (Registre du Cushing – Club français de l’hypophyse) [L'ACTUALITÉ COMMENTÉE] |
• Mifépristone dans le syndrome de Cushing : une étude prospective - Charlène Cordray [MISE AU POINT SUR...] Optimiser le traitement substitutif cortisolique - Frédéric Castinetti [L'IMAGE COMMENTÉE] Microprolactinome, quand les coupes axiales font le diagnostic... |

Du neuf dans la prise en charge médicamenteuse de la maladie de Cushing…
Frédéric Castinetti (Service d'endocrinologie, diabète et maladies métaboliques, hôpital de la Timone, Marseille)
Le traitement de la maladie de Cushing fait appel en première ligne à la chirurgie. Cependant, celle-ci peut se révéler inefficace ou délicate en l'absence d'image évidente d'adénome à l'IRM hypophysaire. Les traitements médicamenteux anticortisoliques ont alors toute leur place : kétoconazole, métyrapone ou mitotane ont démontré au moins en partie leur efficacité, même si leur tolérance est parfois médiocre. Certains ont même proposé de les associer, avec une efficacité franche, dans les hypercorticismes sévères (Kamenicky P et al., J Clin Endocrinol Metab 2012).
Ce numéro 2 de la newsletter Hypophyse fait la part belle à 2 traitements nouveaux ou revisités de la maladie de Cushing :
- Le pasiréotide (Signifor®, laboratoires Novartis), somatostatinergique de forte affinité pour les récepteurs somatostatinergiques sst5, a déjà démontré une efficacité antisécrétoire dans la maladie de Cushing. La première étude de phase III portant sur un nombre conséquent de patients porteurs de maladie de Cushing vous est également rapportée dans cette newsletter (Colao A et al. N Engl J Med 2012). À noter, l'essai thérapeutique en cours sur le pasiréotide à libération prolongée dans la maladie de Cushing, qui permettra de définir le profil de patients le plus à même de bénéficier de ce traitement.
- La mifépristone (Korlym®, laboratoires Corcept Therapeutics), le seul antagoniste des récepteurs aux glucocorticoïdes, qui a désormais une autorisation d'utilisation validée par la Food and Drug Administration dans l'hypercorticisme. La mifépristone a démontré son efficacité clinique dans le traitement des hypercorticismes, mais la plupart des études publiées à ce jour étaient rétrospectives ou portaient sur un faible nombre de sujets (Castinetti F et al., Curr Opin Endocrinol Diabetes Obes 2012). M. Fleseriu et al. rapportent dans le JCEM une étude prospective qui confirme cette efficacité (article résumé dans cette newsletter ; Fleseriu M et al., J Clin Endocrinol Metab 2012).
Le Congrès européen d'endocrinologie a également permis de se familiariser avec le LCI699, un nouvel inhibiteur de la 21-hydroxylase développée par Novartis (communication orale présentée par l'équipe du Pr Bertagna). Le traitement, administré pendant 10 semaines à 11 patients porteurs du syndrome de Cushing, a permis, chez les 9 patients qui ont achevé l'étude, une normalisation du cortisol libre urinaire des 24 heures. Principal effet indésirable – attendu –, la survenue d'une hypokaliémie, chez 4 patients. L'hyperandrogénie, également attendue, n'a pas été rapportée chez les patientes de l'étude, mais l'effectif était restreint.
Nous devons donc nous attendre à un changement de nos pratiques et des recommandations dans les prochaines années. Pour l'heure, et avant la déferlante potentielle de ces nouveaux traitements, une mise au point finale sur l'efficacité des anticortisoliques classiques nous semble nécessaire. Dans cette optique, je me permets de profiter de cet éditorial pour rappeler que nous faisons actuellement, en accord avec les responsables du Registre français du Cushing et du Club de l'hypophyse, une étude rétrospective sur les patients traités par ces anticortisoliques dans le cadre de la maladie de Cushing. Pour ceux parmi vous qui seraient intéressés, vous pouvez télécharger la fiche de recueil de données, et me la renvoyer par mail (frederic.castinetti@ap-hm.fr) ou par courrier (Service d'endocrinologie du Pr Brue, hôpital de la Timone, rue Saint-Pierre, 13005 Marseille).
En vous souhaitant une agréable lecture, et en vous remerciant pour votre intérêt.
PS : La Newsletter Hypophyse ne serait pas ce qu'elle est sans la fameuse IRM que le Pr Bonneville nous fait la joie de nous fournir. À noter, également, une autre nouveauté thérapeutique prometteuse, l'octréotide per os, dont l'étude princeps vous est résumée par le Dr Galland, et un case report sur l'association mutation de SDHD/Acromégalie résumé par le Dr Cazabat.

Le pasiréotide en traitement de la maladie de Cushing : résultats d'une étude de phase III
Juliette du Payrat (Lyon), Gérald Raverot (Lyon)
Colao A, Petersenn S, Newell-Price J. A 12-month phase 3 study of pasireotide in Cushing's disease. N Engl J Med 2012;366:914-24.
Les adénomes corticotropes expriment de façon préférentielle les récepteurs de la somatostatine de sous-type 5. Le pasiréotide (SOM230) est un analogue de la somatostatine qui présente une haute affinité pour les récepteurs de sous-type 5, ce qui en fait un candidat de choix dans le traitement médical des adénomes corticotropes non guéris ou non accessibles à la chirurgie.
Cette publication correspond aux résultats d'un essai de phase III, randomisé et en double aveugle, ayant inclus 162 patients adultes présentant une maladie de Cushing active (définie par un cortisol libre urinaire supérieur à 1,5 fois la valeur supérieure de la normale) : persistante, récurrente ou non accessible à la chirurgie. Les patients ont été répartis en 2 groupes recevant le pasiréotide par voie sous-cutanée, en 2 injections quotidiennes, de 600 µg pour 82 des patients et 900 µg pour les 80 autres. À 3 mois, la dose a été majorée de 300 µg 2 fois par jour jusqu'à 6 mois, chez tous les patients dont le cortisol libre urinaire des 24 heures (CLU) initial excédait 2 fois la limite supérieure de la normale, et chez tous ceux dont le CLU à 3 mois n'était pas normalisé. L'étude a ensuite été poursuivie de façon ouverte jusqu'à 12 mois.
L'objectif principal était l'obtention à 6 mois d'un CLU normalisé sans majoration de dose. Les objectifs secondaires étaient la normalisation du CLU à 3, 6 et 12 mois, quelle qu'ait été l'adaptation de la posologie, le contrôle partiel de l'hypercorticisme (défini par une réduction de 50 % ou plus du CLU), ainsi que l'étude de l'évolution des paramètres cliniques et biologiques liés au syndrome de Cushing.
L'objectif principal a été atteint chez 15 % (12/82) des patients du groupe 600 µg contre 26 % (21/80) du groupe 900 µg. En incluant les patients ayant nécessité une majoration de dose, respectivement 16 % et 29 % des patients étaient contrôlés à 6 mois, et 13 % et 25 % à 12 mois.
La médiane du CLU a diminué rapidement de 50 % à 2 mois et est restée stable par la suite dans les 2 groupes. En revanche, chez les 72 patients non contrôlés précocement à 1 et 2 mois, 92 % restaient non contrôlés à 6 mois et 89 % à 12 mois.
Il a été constaté dans les 2 groupes une amélioration des paramètres cliniques et biologiques à 12 mois : pression artérielle, poids (avec une perte moyenne de 6 kg à 12 mois), qualité de vie, bilan lipidique.
Parmi les 75 patients qui présentaient initialement une tumeur hypophysaire mesurable, il a été observé une réduction tumorale moyenne de 9,5 % dans le groupe 600 µg et de 43,8 % dans le groupe 900 µg.
Concernant la tolérance, 73 % des patients (118 des 162) ont présenté une augmentation rapide de la glycémie après l'instauration du pasiréotide : à 12 mois, 48 % des patients non diabétiques initialement avaient une hémoglobine glyquée supérieure à 6,5 %, et 74 des 162 patients avaient dû recevoir un nouveau traitement antidiabétique. Six pour cent des patients ont interrompu l'étude en raison d'un effet indésirable lié à l'hyperglycémie. Aucun cas d'acidocétose ou de coma hyperosmolaire n'a été décrit. Le reste du profil de tolérance était similaire à celui des autres analogues de la somatostatine.
En conclusion, le pasiréotide induit une réduction significative de l'hypercorticisme et semble avoir sa place dans le traitement des adénomes corticotropes non accessibles à la chirurgie ou non guéris, mais une vigilance particulière doit être accordée à la tolérance glycémique. La réponse précoce au traitement semble être prédictive de la réponse ultérieure.
Mifépristone dans le syndrome de Cushing : une étude prospective
Charlène Cordray (Service d'endocrinologie, diabète et maladies métaboliques, hôpital de la Timone, Marseille)
Fleseriu M, Biller BM, Findling JW et al. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's Syndrome. J Clin Endocrinol Metab 2012 [Epub ahead of print].
La mifépristone est un antagoniste des récepteurs de la progestérone utilisé principalement à visée contragestive. Elle possède, à plus forte dose, une action antagoniste des récepteurs aux glucocorticoïdes. L'objectif de cette étude était d'évaluer l'efficacité clinique et métabolique de la mifépristone en traitement du syndrome de Cushing chez des patients résistants aux autres thérapeutiques. Il s'agit de la première étude prospective portant sur un nombre raisonnable de patients traités par mifépristone pour un syndrome de Cushing (en fait, principalement une maladie de Cushing, portée par plus de 80 % des patients de l'étude).
Cette étude prospective a été réalisée sur 24 semaines dans 17 centres américains. Ont été inclus des patients atteints d'un syndrome de Cushing associé à un diabète et/ou à une intolérance au glucose et/ou à une hypertension artérielle. On rappelle à ce stade que le principe même d'action de la mifépristone ne permet pas d'utiliser les valeurs de cortisol plasmatique ou urinaire comme marqueur d'efficacité. Le critère de jugement principal en termes d'équilibre glycémique était une diminution d'au moins 25 % de l'aire sous la courbe de la réponse glycémique à l'hyperglycémie provoquée par voie orale (HPO). Concernant l'équilibre tensionnel, une baisse d'au moins 5 mmHg de la tension artérielle diastolique (TAD) était considérée comme significative.
Cinquante patients ont été inclus de janvier 2008 à janvier 2011, chaque patient recevant une dose quotidienne de 300 à 1 200 mg de mifépristone. Une diminution significative de l'aire sous la courbe en réponse à l'HPO (p < 0,001), ainsi qu'une baisse significative de l'HbA1c (passée de 7,43 ± 1,52 % à 6,29 ± 0,99 %, p < 0,001) et de la glycémie à jeun (passée de 1,49 ± 0,75 g/l à 1,04 ± 0,37g/l, p < 0,03) ont été mises en évidence chez 60 % des patients diabétiques ou intolérants au glucose (p < 0,00001). Ont également été démontrés une baisse significative de la tension artérielle diastolique chez 38 % des patients (p < 0 ,05), ainsi qu'une diminution significative du poids (- 5,7 ± 7,4 %, p < 0,001) et du tour de taille. Ce taux médiocre d'amélioration des chiffres tensionnels s'explique probablement par une fixation du cortisol en excès sur les récepteurs minéralocorticoïdes, non bloqués sous mifépristone. Enfin, la majorité des patients ont présenté une amélioration des scores de qualité de vie.
Les effets indésirables survenus pendant l'étude sont aspécifiques : asthénie, nausées et céphalées. Comme décrit précédemment, la mifépristone a occasionné des hypokaliémies (contrôlables sous potassium et anti-aldostérone), et parfois des augmentations des chiffres tensionnels. Enfin, des métrorragies (hyperœstrogénie) ont également été rapportées.
Les auteurs ont donc réussi à démontrer l'efficacité de la mifépristone en tant que traitement du syndrome de Cushing. L'efficacité de cette molécule ne pouvant être évaluée sur la cortisolémie (taux augmentés du fait du blocage des récepteurs aux glucocorticoïdes), les auteurs ont réussi, avec des critères de jugement pertinents et objectifs, à prouver que la mifépristone améliore significativement les troubles cliniques et métaboliques du syndrome de Cushing. Ce traitement étant désormais autorisé aux États-Unis dans l'indication d'hypercorticisme, il est probable que nous le reverrons prochainement en Europe dans cette indication. Pour rappel, l'un de ses principaux avantages est sa rapidité d'action, qui en fait un traitement de choc, par exemple dans les formes psychiatriques.
Première étude chez l'homme d'une forme orale d'octréotide
Françoise Galland (Service d'endocrinologie et de diabétologie, CHU de Rennes)
Tuvia S, Atsmon J, Teichman SL et al. Oral octreotide absorption in human subjects: comparable pharmacokinetics to parenteral octreotide and effective growth hormone suppression. J Clin Endocrinol Metab 2012;97(7) [Epub ahead of print].
L'attente d'une forme orale d'analogue de la somatostatine est extrêmement importante car elle permettrait d'améliorer, entre autres, la qualité de vie des patients. La difficulté jusqu'à présent était de développer une forme orale capable de franchir la barrière intestinale. Cette étude est la première chez l'homme. L'objectif des auteurs était d'étudier, d'une part, la pharmacocinétique d'une préparation d'octréotide oral, d'autre part, son action sur le taux de GH chez 75 volontaires sains, en comparaison à l'octréotide sous-cutané (s.c.). Cette forme orale d'octréotide est une préparation à base d'Octreolin® (Chiasma, Jérusalem, Israël) mis en suspension dans une capsule qui a la propriété d'entraîner une perméabilité transitoire de l'intestin et ainsi de laisser passer la forme active de l'octréotide qui est à l'intérieur. Les posologies testées étaient pour l'octréotide s.c. : 0,1 mg en 1 seule fois et pour l'octréotide oral : 3, 10 et 20 mg.
Résultats sur les paramètres pharmacocinétiques : il est mis en évidence une relation dose-concentration d'octréotide plasmatique pour les 3 posologies de la forme orale ; la présence de l'octréotide dans le plasma est mise en évidence 1 heure après l'administration orale ; le profil pharmacocinétique après 20 mg oral est équivalent à celui après 0,1 mg s.c. : pic moyen de concentration plasmatique : 3,77 ± 0,25 versus 3,97 ± 0,19 ng/ml respectivement ; délai moyen pour une concentration thérapeutique ≥ 0,5 ng/ml : 7,67 versus 5,88 heures, respectivement ; la prise orale concomitante à un repas réduit la biodisponibilité de l'octréotide de 90 % ; cela peut être atténué (biodisponibilité diminuée de 40 %) par la prise d'inhibiteur de la pompe à protons préalablement.
Résultats sur l'action antisécrétoire (sécrétion de GH) : après une prise unique de 20 mg d'octréotide oral, les auteurs ont mis en évidence une diminution de GH basale de 49 % (p < 0,05) et de 80 % après stimulation GHRH (p < 0,001).
Tolérance : la forme orale est bien tolérée : effets indésirables digestifs (douleur abdominale dans 15 % des cas, diarrhée dans 8 % des cas, nausées dans 7 % des cas, selles décolorées dans 10 % des cas), céphalées chez 10 % des patients. Pas de différence significative avec la forme s.c.
En conclusion, ces résultats sont très prometteurs ; on attend maintenant les études chez des patients acromégales.
SDHs : nouveaux gènes de prédisposition aux adénomes hypophysaires ?
Laure Cazabat (Service d'endocrinologie et de diabétologie, hôpital Ambroise-Paré, AP-HP)
Xekouki P, Pacak K, Almeida M et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: a new association for SDH? J Clin Endocrinol Metab 2012;97:E357-66.
Depuis la découverte de mutations de SDHD en 2000 (1), les autres sous-unités SDHB, SDHC et, plus récemment, SDHA, ont été impliquées dans les phéochromocytomes/paragangliomes (PHEO/PGL) familiaux. Ces mutations des SDH ont été décrites depuis dans d'autres tumeurs : le GIST (gastrointestinal stromal tumor), la dyade de Carney-Stratakis (GIST et PGL), le cancer du rein, le syndrome de Cowden PTEN-négatif et de Cowden-like, le cancer papillaire de la thyroïde, etc. (2).
Les auteurs décrivent dans cet article le cas d'une association acromégalie et PGL chez un patient avec une mutation du gène SDHD. Ces mutations SDHD sont classiquement associées aux PGL familiaux, principalement de la tête et du cou, avec fréquemment de multiples localisations (80 %) ; ils sont exceptionnellement malins (< 5 %).
L'acromégalie est diagnostiquée à l'âge de 37 ans (IGF1 2N) avec, à l'IRM, un macroadénome somatotrope invasif de 35 mm (GHRH normal). Elle est associée à une hypertension artérielle : les normétanéphrines urinaires sont à 8N. L'exploration morphologique retrouve de multiples localisations de PGL/PHEO (5 PGL de la tête et du cou, 5 thoraciques et rétropéritonéaux, et des PHEO bilatéraux). Une surrénalectomie bilatérale ne permet pas le contrôle de la sécrétion (normétanéphrines urinaires à 5N). L'acromégalie n'est pas contrôlée par les agonistes de la somatostatine, et le patient est opéré par voie transsphénoïdale ; en postopératoire, l'IGF1 est normalisée sous agonistes. Étonnamment, les métanéphrines urinaires diminuent après la chirurgie hypophysaire (de 5N à 2N), sans modification en taille des tumeurs connues.
Dans la famille, il y a un antécédent de PGL chez un oncle, mais aucun cas d'adénome hypophysaire. L'étude génétique retrouve une mutation du gène SDHD conduisant à un décalage du cadre de lecture et un codon stop précoce : pour la tumeur hypophysaire, aucune anomalie sur les gènes AIP, MEN1 et CDKN1B.
Dans la littérature, on retrouve 2 autres cas d'adénome hypophysaire chez des patients avec mutation de SHDs (SDHB type d'adénome chez un patient avec phéochromocytome à 15 ans (3) et SDHC, macroprolactinome à l'âge de 60 ans [4]), sur plus de 700 patients… (2). Quant à l'association PHEO ou PGL et adénome hypophysaire, 25 cas ont été rapportés dans une revue de la littérature en 2003 (5) et au moins 4 depuis (6, 7), dont 2 NEM2a (8, 9), le plus fréquemment des adénomes somatotropes.
Les auteurs ont cherché des éléments en faveur du rôle de cette mutation SDHD dans le développement de la tumeur somatotrope. Ils montrent que la protéine SDHD est exprimée dans l'hypophyse normale et un adénome somatotrope non muté SDHs (contrôle), mais pas dans la tumeur du patient : ils constatent une perte d'hétérozygotie de SDHD et une absence de la protéine SDHD ; les SDHs sont connus pour avoir un rôle suppresseur de tumeur. De même, HIF1-α est augmenté dans la tumeur SDHD par rapport au contrôle ; cette augmentation est liée à un état de pseudohypoxie dans les tumeurs avec mutations de SDHs, et HIF1-α est connu pour activer la tumorigenèse.
D'autre part, ils retrouvent pour la première fois une expression de GHR dans les phéochromocytomes du patient et d'autres phéochromocytomes avec mutations de SDHs, ce qui peut expliquer la diminution de la sécrétion de catécholamines après traitement de l'acromégalie par le biais de la diminution de GH.
C'est la première description de l'étude des conséquences d'une mutation de SDHD dans un adénome hypophysaire chez un patient avec PGL familiaux. Néanmoins, le rôle de cette mutation dans la tumorigenèse hypophysaire reste à démontrer. La rareté des patients porteurs de mutations SDHs chez lesquels un adénome hypophysaire est retrouvé fait rester très prudent sur le rôle des gènes SDHs comme gènes de prédisposition aux adénomes hypophysaires.
Références bibliographiques
1. Baysal BE, Ferrell RE, Willett-Brozick JE et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000;287:848-51.
2. Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009;266:19-42.
3. Benn DE, Gimenez-Roqueplo AP, Reilly JR et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006;91:827-36.
4. Lopez-Jimenez E, de Campos JM, Kusak EM et al. SDHC mutation in an elderly patient without familial antecedents. Clin Endocrinol (Oxf) 2008;69:906-10.
5. Breckenridge SM, Hamrahian AH, Faiman C, Suh J, Prayson R, Mayberg M. Coexistence of a pituitary macroadenoma and pheochromocytoma – a case report and review of the literature. Pituitary 2003;6:221-5.
6. Sisson JC, Giordano TJ, Avram AM. Three endocrine neoplasms: an unusual combination of pheochromocytoma, pituitary adenoma, and papillary thyroid carcinoma. Thyroid 2012;22:430-6.
7. Zhang C, Ma G, Liu X et al. Primary cardiac pheochromocytoma with multiple endocrine neoplasia. J Cancer Res Clin Oncol 2011;137:1289-91.
8. Heinlen JE, Buethe DD, Culkin DJ, Slobodov G. Multiple endocrine neoplasia 2a presenting with pheochromocytoma and pituitary macroadenoma. ISRN Oncol 2011;2011:732452.
9. Saito T, Miura D, Taguchi M, Takeshita A, Miyakawa M, Takeuchi Y. Coincidence of multiple endocrine neoplasia type 2A with acromegaly. Am J Med Sci 2010;340:329-31.

Optimiser le traitement substitutif cortisolique
Frédéric Castinetti (Service d'endocrinologie, diabète et maladies métaboliques, hôpital de la Timone, Marseille)
Le déficit corticotrope est une pathologie rare, en général provoquée par une tumeur hypothalamo-hypophysaire, ou par son traitement (déficit corticotrope induit par la chirurgie ou la radiothérapie). Sa prévalence est estimée à 3 pour 10 000 personnes, ce qui la rend plus fréquente que l'insuffisance surrénalienne primaire. Le traitement de référence fait appel à l'hydrocortisone à la dose théorique de 15 à 20 mg/jour : le schéma d'administration (heure et posologie à chaque prise) n'est cependant pas consensuel, et peut comporter 2 à 3 prises à des heures variables de la journée. La surveillance est clinique, fondée sur l'état général et les signes fonctionnels du patient, le poids et la tension artérielle.
Il existe plusieurs inconvénients à la prise en charge thérapeutique actuelle par hydrocortisone :
- Les schémas de prise ne miment pas la sécrétion circadienne du cortisol, ce qui entraîne des alternances de sur- et de sous-dosage au cours des 24 heures, et un profil en pic au cours de la journée. Un sous-dosage en hydrocortisone se manifeste par une asthénie chronique dès le lever, une hypotension artérielle, avec altération de la qualité de vie, des troubles comportementaux et une augmentation globale de la morbimortalité.
- La dose quotidienne totale d'hydrocortisone est en général trop importante. Un surdosage en hydrocortisone peut être à l'origine de perturbations cliniques (prise de poids et modification de la composition corporelle, hypertension artérielle), métaboliques (dyslipidémie, hyperglycémie) et osseuses. Un surdosage entraîne une surmortalité en comparaison avec la population générale et une altération de la qualité de vie. Ce surdosage chronique modéré est cependant souvent délicat à diagnostiquer, au moins à court terme.
- L'observance est parfois imparfaite, en particulier chez les sujets jeunes, du fait de la nécessité de plusieurs prises par jour.
En 2010, nous avions mené une étude d'évaluation pharmacocinétique (Simon N, Castinetti F et al., Clin Pharmacokinet 2010), incluant 50 patients, dont 30 avec un déficit corticotrope. La mesure des taux de cortisol plasmatique toutes les 2 heures pendant 24 heures, avec prise du traitement habituel par hydrocortisone, nous avait permis de définir un modèle de simulation pharmacocinétique. Nous avions ensuite simulé 18 différents schémas de prise d'hydrocortisone, et nous avions ainsi pu déterminer le pourcentage de patients qui atteignait les cibles thérapeutiques biologiques de cortisolémie à 8 heures (30 minutes après la prise), à 16 heures et à minuit. La simulation de plus de 500 patients avec différents schémas de prise a montré que même le meilleur schéma (3 prises d'hydrocortisone, avec 10 mg à 7 h 30, 5 mg à midi, et 5 mg à 16 h 30) ne permettait pas d'obtenir plus de 50 % de patients dans les zones biologiques thérapeutiques, la majorité des patients étant sous- ou surdosés aux divers moments de la journée.
Dans ce contexte de traitement substitutif imparfait, des études ont été menées sur de nouvelles formes d'hydrocortisone à libération modifiée. Le Plenadren® (anciennement dénommé Duocort®) est ainsi une forme d'hydrocortisone à double libération, qui existe en 2 dosages, à 5 et 20 mg. Le comprimé comprend 1 partie à libération immédiate (enrobage), dont le pic plasmatique est atteint 20 à 40 minutes après la prise, et 1 partie à libération prolongée (noyau), dont la concentration plasmatique diminue progressivement et continuellement en 12 à 16 heures après la prise. Aucune libération nocturne de cortisol n'a été observée. Le profil est identique pour les 2 dosages de comprimés à 5 et à 20 mg (Johannsson G, Filipsson H et al., Horm Res 2007). Le traitement se prend en 1 prise unique le matin à jeun. Le Plenadren® a obtenu la désignation orpheline dans l'Union Européenne dans l'indication "traitement de l'insuffisance surrénalienne" en 2006, et a une AMM européenne depuis novembre 2011 (EMEA/H/C/002185).
G. Johannsson et al. ont récemment rapporté la première étude comparative sur le Plenadren®. Cette étude prospective multicentrique randomisée portait sur 64 patients atteints d'insuffisance surrénalienne exclusivement primaire. Les patients étaient divisés en 2 groupes : le 1er groupe recevait le Plenadren® à la dose totale administrée habituellement avec leur hydrocortisone, pendant que le 2e groupe conservait un traitement par hydrocortisone, administré en 3 prises (moitié de dose à 8 heures, quart de dose à midi, quart de dose à 16 heures), à sa dose habituelle. L'essai était de type croisé : après 3 mois du traitement initial, chaque groupe bénéficiait de l'autre thérapeutique pendant 3 mois. Le critère principal était la comparaison de la biodisponibilité ; les critères secondaires évaluaient la tolérance, l'efficacité et la qualité de vie. L'étude pharmacocinétique a souligné que Plenadren® permettait d'augmenter la biodisponibilité d'hydrocortisone pendant les 4 premières heures et de la diminuer entre la 4e et la 24e heure, avec 1 seul véritable pic de survenue rapide après la prise. Pour les critères secondaires, les patients traités par Plenadren® ont amélioré significativement leurs paramètres métaboliques (poids, pression artérielle systolique et diastolique, HbA1c) en comparaison avec le traitement classique. Les index de qualité de vie ont montré soit un score inchangé (échelle SF36), soit une amélioration du score (échelle de fatigue). Les effets indésirables ont été comparables en fréquence dans les 2 groupes : aucun effet indésirable grave n'a été rapporté. La majorité des patients a présenté une amélioration des scores de qualité de vie, et 92 % ont accepté de poursuivre l'étude dans le cadre d'une première phase d'extension de 6 mois, puis d'une deuxième phase d'extension de 18 mois. L'ensemble des résultats observés à court terme a été retrouvé après un suivi total de 27 mois (données présentées à l'ECE 2012). Cette étude souligne donc, dans une population d'insuffisants surrénaliens primaires, l'intérêt d'un traitement par Plenadren® en remplacement du schéma classique d'hydrocortisone (Johannsson G, Nilsson AG et al., J Clin Endocrinol Metab 2012). Il n'existe pas à ce jour de données sur le Plenadren® dans le déficit corticotrope, même s'il est probable que l'efficacité soit comparable.

Microprolactinome, quand les coupes axiales font le diagnostic...
Jean-François Bonneville (CHU de Besançon)
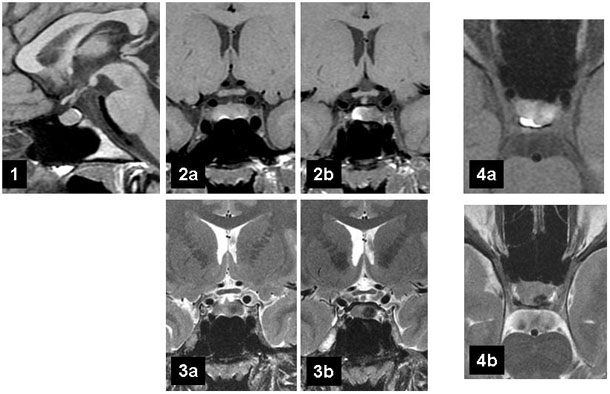
Juliette, 17 ans, consulte avec sa mère pour une galactorrhée isolée bilatérale. Ses premières règles sont survenues à 16 ans, avec des cycles longs. À l'interrogatoire, elle dit avoir eu un épisode de céphalées quelques mois auparavant. Son taux de prolactine est à 1 096 mUI/l.
Il n'y a pas de contraception hormonale et pas de cause iatrogène.
Devant cette galactorrhée isolée avec un taux de prolactine peu élevé, le diagnostic clinique n'est pas clair.
En IRM, la séquence sagittale T1 (figure 1) est normale – comme c'est souvent le cas dans les lésions purement intrasellaires.
Les coupes coronales T1 et coronales T2 passant par le centre de la selle turcique (figures 2a et 3a), elles ne sont pas vraiment informatives.
Les coupes T1 et T2 plus postérieures (figures 2b et 3b) mettent en évidence une asymétrie du contenu sellaire, mais on pourrait ici discuter :
- en T1 : une latéralisation physiologique du lobe postérieur (dans 50 % des cas, le lobe postérieur n’est pas médian) ;
- en T2 : une image de volume partiel avec le dos de la selle turcique ou avec une expansion du sinus sphénoïdal.
En cas de difficulté diagnostique, plutôt que de recourir systématiquement aux séquences après injection de gadolinium, nous privilégions les séquences axiales.
- La coupe axiale T1 (figure 4a) met en évidence de façon formelle un aspect de compression du lobe postérieur par une lésion intrasellaire latérale gauche infracentimétrique, hétérogène, dont le centre est en discret hypersignal.
- La coupe axiale T2 révèle à la partie postérieure de cette lésion une image lenticulaire fortement hypo-intense, signant le caractère hémorragique de la lésion.
Au total, l'aspect IRM est tout à fait compatible avec un prolactinome avec transformation hémorragique partielle, ce qui est cohérent avec un tableau clinique fruste et un taux de prolactine très faiblement augmenté. Une réduction de volume de la lésion après traitement par agonistes de la dopamine confirmera le diagnostic.
